






蛋白质的数据预处理个人笔记(2)
相互作用残基对的定义
两个蛋白质的两个残基之间的欧几里德距离小于等于6 Å(埃格斯特朗,1 Å = 0.1奈米),则认为这两个残基之间有相互作用。根据这个定义,得到12138个阳性样本(相互作用的残基对)和5,522,852个阴性样本(非相互作用的残基对)
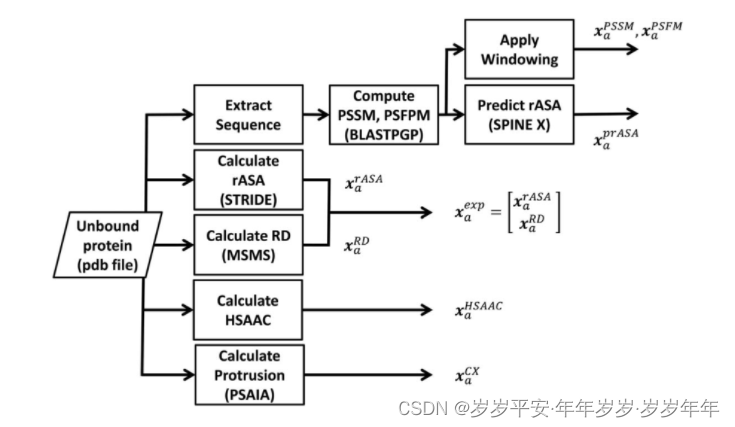
相对可及表面积(rASA)、残基的深度、突出指数、疏水性和半球氨基酸组成。最后,我们将上述所有特征结合起来,得到70维的节点特征。边的特征由平均原子距离和两个残基之间的夹角组成
节点特征
-
相对可及表面积(rASA):使用STRIDE计算给定蛋白质结构的相对可及表面积(rASA)(STRIDE:基于原子坐标的蛋白质二级结构自动分配算法),范德瓦尔斯作用面的表面积,实际操作中会用一个小球通过范德瓦尔斯作用在残基附近滚动一遍,取球心包络面的面积
-
残基深度RD:残基深度定义为残基到蛋白质表面的最小距离,并且使用MSMS计算。MSMS将残基深度值归一化。rASA和RD 连接形成单一的表面曝光特征向量,用x_a表示。我们发现残基深度在残基相互作用预测中具有与rASA中互补的信息
-
半球形氨基酸组成(HSAAC):这个特征捕获了残基侧链和侧链相反方向上的氨基酸组成。某一方向上的氨基酸组成定义为特定氨基酸在该方向上与感兴趣的残基8.0 a˚的最小原子距离阈值内出现的次数。因此,HSAAC结合了表面可及性和残基附近的氨基酸组成。
将这两个方向的氨基酸组成向量归一化得到单位范数,得到HSAAC-u和HSAAC-d,再将两者串联得到HSAAC。我们利用Biopython 计算HSAAC。
-
突出指数(CX):非氢原子的突出指数被定义为半径为10.0 a˚、以该原子为中心的球体的体积占该原子的体积的比例.单残基的突出指数是一个六维向量,由残基中所有原子的突出值的平均值、标准差、最大值和最小值以及其侧链原子的突出值的平均值和标准差组成。这个向量的每个元素都被归一化,其范围从0到1。
-
疏水性:PSAIA作为工具,得到的疏水性程度会被Normalize到0~1上
边的特征
-
平均原子距离:特征数:1(仅标量距离),两个残基各原子的平均距离。距离计算出来以后会被高斯函数挤压到0~1
-
两个残基之间的夹角:对一个残基,根据其alpha碳、羧基碳、羧基氧的位置可以定义出一个单位向量。于是对两个残基,可以由此定义出方位角,最后再/2Pi化到0~1即可
位置特异性打分矩阵(PSSM)和位置特定频率矩阵(PSFM)
PSSM通过序列相似性比对生成,将氨基酸的保守性信息包含在矩阵中
位置特定评分矩阵( PSSM)和位置特定频率矩阵(PSFM)反映了基于进化信息的蛋白质链特定位置残基的保守性,采用滑动窗口的方法用PSIBLAST对NCBI NR数据库进行3次迭代,e值设置为0.001得到PSSM和PSFM,最后得到一个3 × 40的矩阵
来自博客:https://blog.csdn.net/CBB_FT/article/details/124623766
- 生成PSSM矩阵
生成序列文件
在UniPort网页中搜索蛋白质名称,并从中下载包含序列等信息的fasta文件,只读取其中的序列并生成单独的序列文件。(在使用BLAST软件生成PSSM时,文件中只能包含一条序列)
下载与安装BLAST
构建BLAST本地数据库
生成PSSM
只能包含一条序列)
下载与安装BLAST
构建BLAST本地数据库
生成PSSM
Python实现批量生成PSSM矩阵















 2175
2175











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









