





论文题目:ResNetKhib: a novel cell type-specific tool for predicting lysine 2-hydroxyisobutylation sites via transfer learning
期刊:Briefings in Bioinformatics
论文链接:https://doi.org/10.1093/bib/bbad063
背景
赖氨酸2-羟基异丁基化(Khib)是最重要的可逆蛋白质翻译后修饰(PTMs)之一,最初由Dai等人在雄性生殖细胞的组蛋白上发现。
尽管现有方法的性能总体良好,但由于以下三个方面,仍存在研究空白,迫切需要开发新方法:(1)先前的研究表明,Khib位点周围的特征在不同物种和细胞类型之间存在差异。然而,目前可用的工具很少可以用于预测细胞类型特异性Khib位点;(2) 最新实验数据集的积累使我们能够开发出更高精度的新方法,这些方法可以应用于蛋白质组学规模上新的Khib位点的鉴定;(3) 目前还没有小鼠Khib位点识别的预测因子。
在此,我们提出了一种名为ResNetKhib的预测因子,旨在准确识别细胞类型特异性的Khib位点。通过设计和应用基于一维卷积框架的ResNet残差连接和基于迁移学习的训练策略。
数据
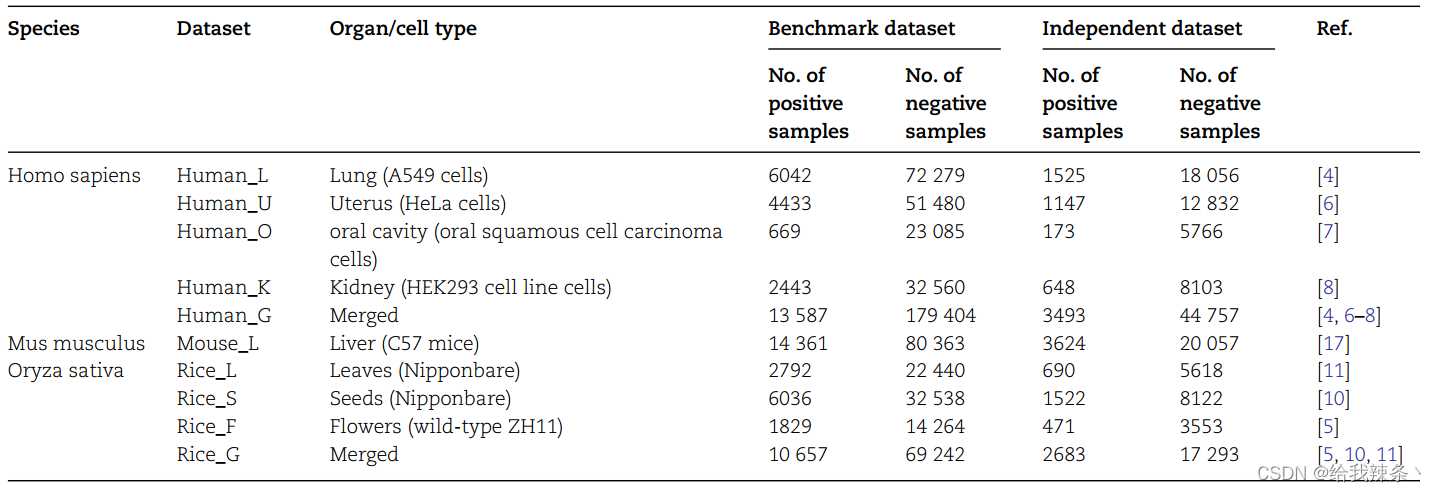
在这项研究中,我们通过搜索文献收集了实验验证的Khib位点,并根据其物种和细胞类型构建了基准数据集。一共收集了10个数据集,包括8个细胞类型特异性数据集和2个一般性数据集。表1提供了基准测试和独立数据集的详细摘要。
表1.基准和独立数据集的统计摘要
为了开发一种用于Khib站点预测的有效机器学习方法,我们采用并评估了多特征编码方案和机器学习算法的性能。使用八种特征编码方案和单词嵌入方法来表示序列。采用随机森林(RF)和基于一维的ResNet算法来集成这些编码。由于带有单词嵌入的ResNet具有出色的性能,我们选择它作为开发ResNetKhib预测器的最终模型。此外,我们还分析了不同物种和细胞类型的Khib和非Khib位点之间的序列基序保守性,以及一个物种的细胞类型特异性基序。我们比较了我们的方法和最先进的预测器的性能,并实现了ResNetKhib算法的web服务器。
收集了三个物种的实验验证的Khib位点,包括智人(人类)、Mus musculus(小鼠)和水稻(水稻),以构建基准数据集。考虑到Khib位点周围不同细胞类型的氨基酸使用偏差,我们根据其细胞类型构建了数据集。总共提取了8个细胞类型特异性数据集和2个通用数据集。智人的四个细胞类型特异性数据集分别来自肺细胞(A549细胞)、子宫细胞(HeLa细胞)、口腔细胞(口腔鳞状细胞癌细胞)和肾细胞(HEK293细胞);而水稻的三个细胞类型特异性数据集分别来自叶片(日本)、种子(日本)和花(ZH11)。对于M.musculus数据,只有一个细胞类型特异性数据集,来自肝细胞(C57小鼠)
特征编码
CKSAAP
CKSAAP特征编码计算由任何k个残基分离的氨基酸对的频率。以k=0为例,存在400个0-间隔的残基对(即AA、AC、AD、…、YY)。例如,如果残基对AA在长度为L的肽中出现m次,则残基对AA的组成等于m除以肽中0间隔残基对的总数,即m/(L–k+1)。在本研究中,最大k值被设置为3,这产生了1600维(400×4)的特征向量。
EAAC
EAAC编码是基于AAC(氨基酸组成)编码的改进版本,已广泛用于预测多个PTM修饰位点。
它在固定长度的窗口中计算AAC,从每个肽的N-末端连续滑动到C-末端。以下方程式用于计算特征向量

其中N(t,win)是滑动窗口win中t型氨基酸的数量,并且N(win)是该滑动窗口win的肽序列的长度。因此,对于5的滑动窗口大小,具有37个残基的肽对应于33个(=37–5+1)滑动窗口,其特征向量维度为33×20(氨基酸)=480。
EGAAC
EGAAC编码基于EAAC编码,其中20种氨基酸类型根据其物理化学性质分为五大类,包括脂肪族基团(“GAVLMI”)、芳香族基团(“FYW”)、正电荷基团(“KRH”)、负电荷基团(‘DE’)和不带电基团(“TCPNQ”)。针对每个滑动窗口计算每组的频率。因此,对于5的滑动窗口大小,具有37个残基的样本对应于33个(37–5+1)滑动窗口,其特征向量维数为33×5(氨基酸组)=165。
AAindex
AAindex是一个代表氨基酸各种物理化学和生物化学性质的数字指数数据库。从AAindex数据库中收集了566个物理和化学特性。去除缺失值的物理化学性质后,保留了553个物理化学性质。我们使用RF分类器计算了每个属性的性能,并选择了前50个属性。
因此,将具有37个残基的肽转化为37×50(性质)=1850的特征载体。
AAF
使用多元统计分析,AAIndex数据库中氨基酸的理化和生化特性可以转换为五个多维属性,分别选择极性、二级结构、分子量、密码子多样性和静电荷。因此,37个残基的肽可以被编码到185维(37×5)的载体中。
BLOSUM62
BLOSUM62矩阵[24]用于表示蛋白质一级序列信息。包含m×L个元素的基质表示肽,其中L表示肽长度,m=21,这些元素包含21个氨基酸,包括间隙(X)。BLOSUM62矩阵中的每一行用于编码21个氨基酸中的一个。因此,BLOSUM62编码的维数为37×21=777。
Z-scales
对于该描述符,每个氨基酸由Sandberg等人在1998年提出的五个物理化学描述符变量表征。因此,具有37个残基的肽可以被编码到185维(37×5)的载体中。
One-hot
One-hot编码方案是将蛋白质序列转换为数字矢量的最流行和最简单的编码方法。在一个热编码中,每个氨基酸由21维二元载体表示,例如:
A由(100000000000000000000)编码,C由(010000000000000000000)编码
RF
RF本质上是建立在训练数据的N个随机子集上的多个决策树的集合,并且经常报告平均预测性能。在本研究中,实现了基于的RF分类器,决策树的数量设置为300。
ResNetKhib
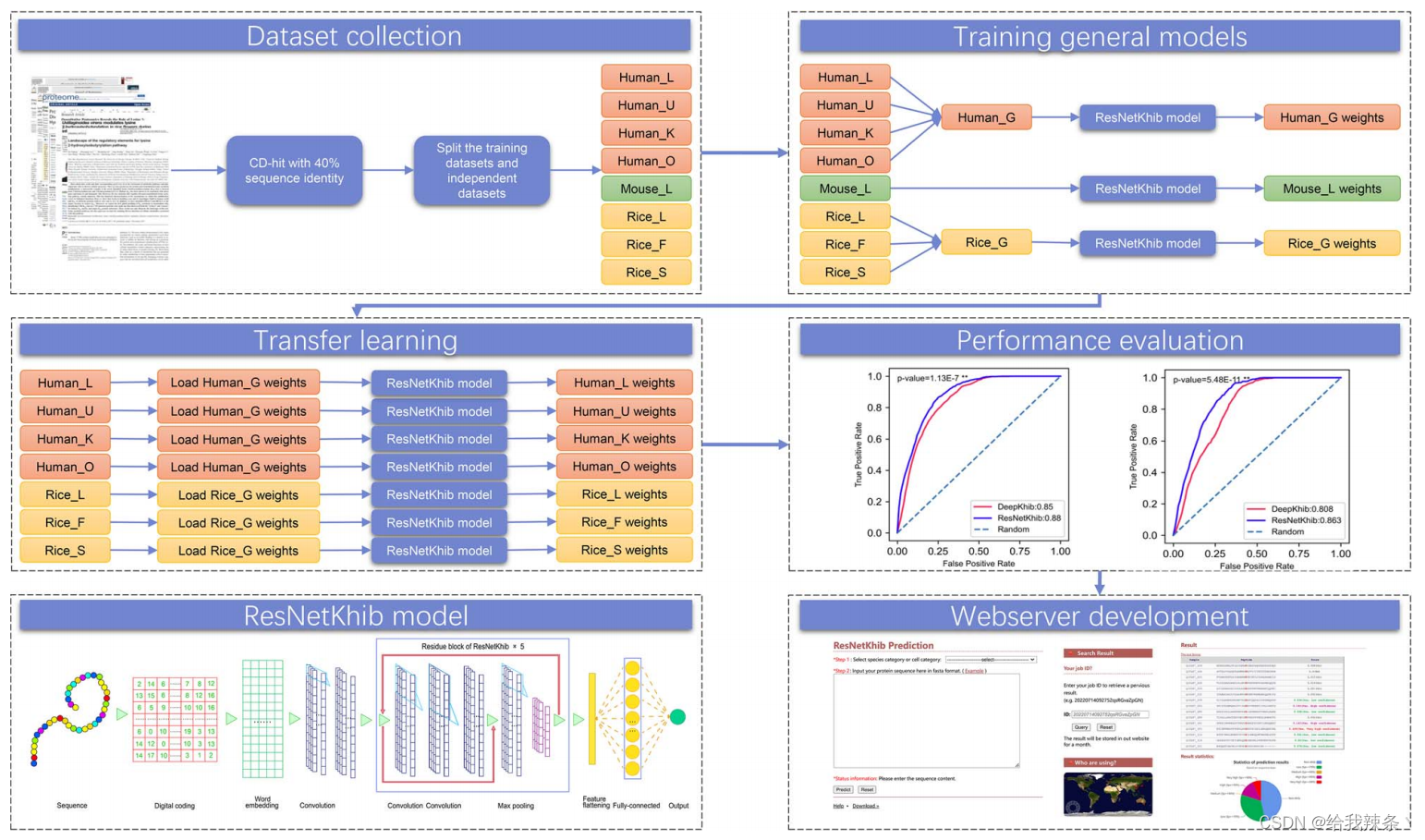
图1. ResNetKhib方法概述
The ResNetKhib model with one-hot encoding
具有一个热编码的ResNetKhib模型(ResNetKhibone热)包含四个模块(补充图S1)。37个残基肽的一个热编码被用作输入层的输入,并且在该模型中没有嵌入层。
卷积模块包含六个卷积块。第一个块是具有64个内核大小为5的滤波器的卷积层,其余模块与ResNetKhibWE中的相应模块相同。
The ResNetKhib model with word embedding
带有字嵌入的ResNetKhib架构(ResNet-KibWE)包括以下五个模块(图2A):
1. 输入层:在该层中,每个具有37个残基的肽被转换为长度为37的索引列表;
2. 嵌入层:将每个索引转换为512维的词向量,以表示氨基酸的性质;
3. 卷积模块:卷积模块包含六个卷积块。第一个块是具有64个核大小为1的滤波器的卷积层,而其余五个是顺序连接的基本残差块(图2B);
4. 全连接层:该层获取上述层的输出,对其进行处理并将其转换为一维向量,该向量包括16个神经元;
5. 输出层:该层仅包含一个神经元,输出最终的概率得分,指示中心赖氨酸残基被2-羟基异丁酰化的可能性。“sigmoid”函数被用作激活函数。
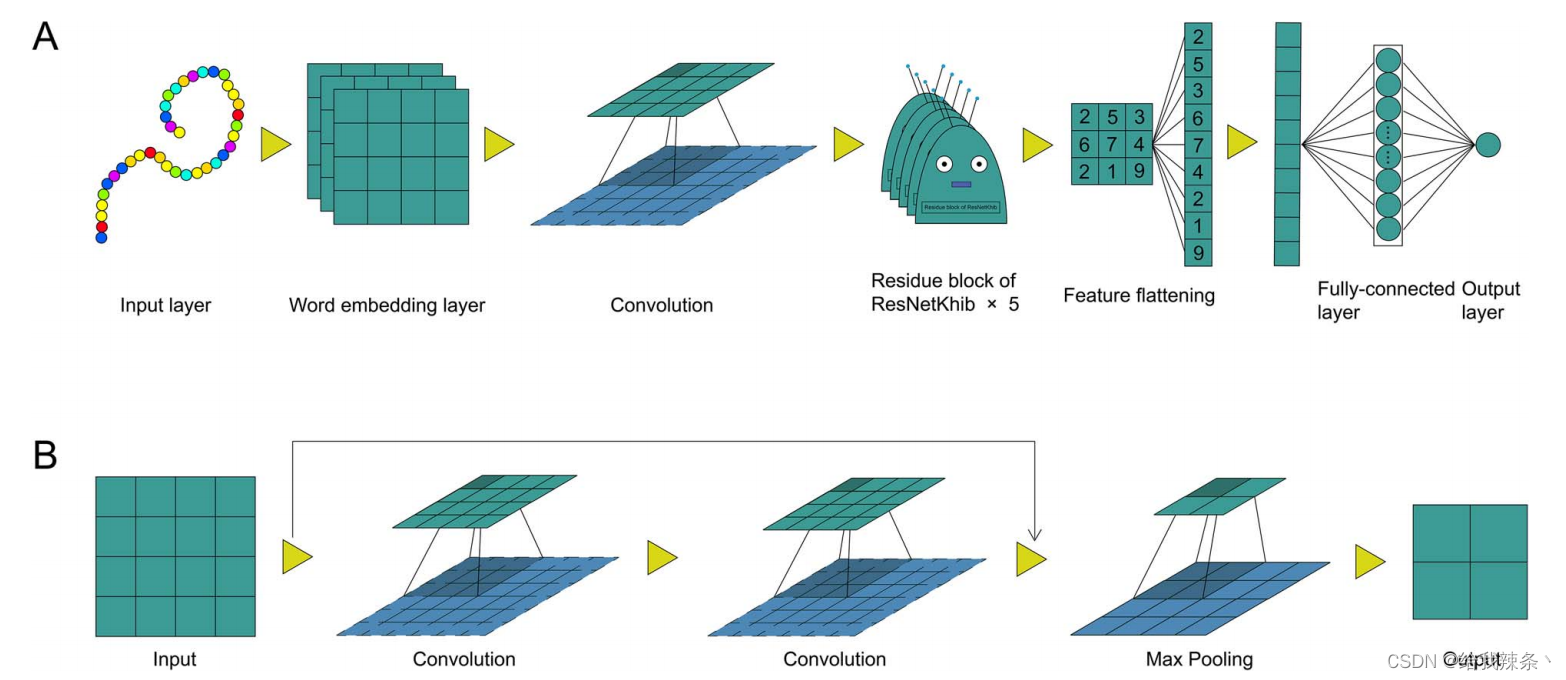
图2. 所提出的ResNetKhib框架的架构(A)和ResNetKhib的残差块(B)
Model training strategy
在本研究中,基于二进制交叉熵(BCE)损失函数,使用Adam算法对一般数据集(即表1中的human_G和rice_G数据集)和Mouse_L数据集的ResNetKhib参数进行了训练和优化。
最大训练周期被设置为1000个时期,以确保损失函数值收敛。在训练过程中也使用了早期停止策略(即,当损失值在100个时期内没有减少时停止训练)。训练数据集在每个历元中被分离,批量大小为1024。
为了避免过拟合,我们在ResNetKhibWE和ResNetKhibone hot的每个卷积层之后添加了dropout操作,dropout率设置为0.5。
BCE损失定义如下:
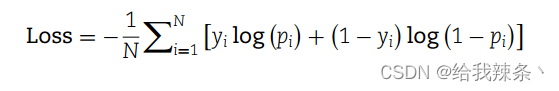
其中N是一批样本的数量,y表示样本的基本事实标签,p分别表示模型的预测值。
细胞类型特定模型的迁移学习
迁移学习是一种机器学习技术,指的是在新问题上重用预先训练的模型,可以通过利用现有知识从源任务到数据有限的目标任务来解决数据稀缺问题。它可以帮助解决数据有限的机器学习任务,提高模型性能。在这项研究中,我们应用迁移学习策略来训练人类和水稻的细胞类型特异性模型。以Human_O模型的训练过程为例,使用训练有素的Human_G模型作为预训练模型。我们首先将预训练模型的参数加载到ResNetKhib网络,然后在没有参数约束的情况下实现微调技术。迁移学习过程也使用Adam算法基于BCE损失函数进行了训练和优化,学习率设置为0.01。
最大训练周期设置为500个时期,并采用早期停止策略来避免过拟合。每个数据集的损失精度曲线如补充图S2所示。
绩效评估策略

结果与讨论
10倍交叉验证和独立测试的性能评估
在本节中,我们首先评估了使用RF分类器的八种不同编码方案的预测性能,方法是进行10倍交叉验证和独立测试(材料和方法部分)。然后,我们比较了基于RF的模型和基于ResNet的模型的性能。补充表S1–S10显示了基于10倍交叉验证和独立测试的10个数据集上不同编码方案在准确性、敏感性、特异性、MCC和AUROC值方面的性能比较。我们可以看到,在基于RF的模型中,AAindex、EAAC和EGAAC编码在10个数据集上的表现明显优于其他编码方案。
以human_U数据集为例(补充表S2),EGAAC编码在10倍交叉验证中获得了最佳性能,AUROC值为0.789(Acc=87.72%;Sn=37.63%;Sp=90.45%;MCC=0.20)。其次是EAAC编码(AUROC=0.783;Acc=87.52;Sn=39.21%;Sp=90.15%;MCC=0.21)和AAindex编码(AUROC=0.766;Acc=87.48%;Sn=39.92%;Sp=9.06%;MCC=0.021)。独立试验的结果与10倍交叉验证试验的结果一致。然后,我们使用相同的策略来评估基于ResNet的模型和性能最好的前三个基于RF的模型的性能(表2)。
表2. 基于10倍交叉验证和独立测试的10个数据集上基于RF的模型和基于ResNet的模型的性能比较
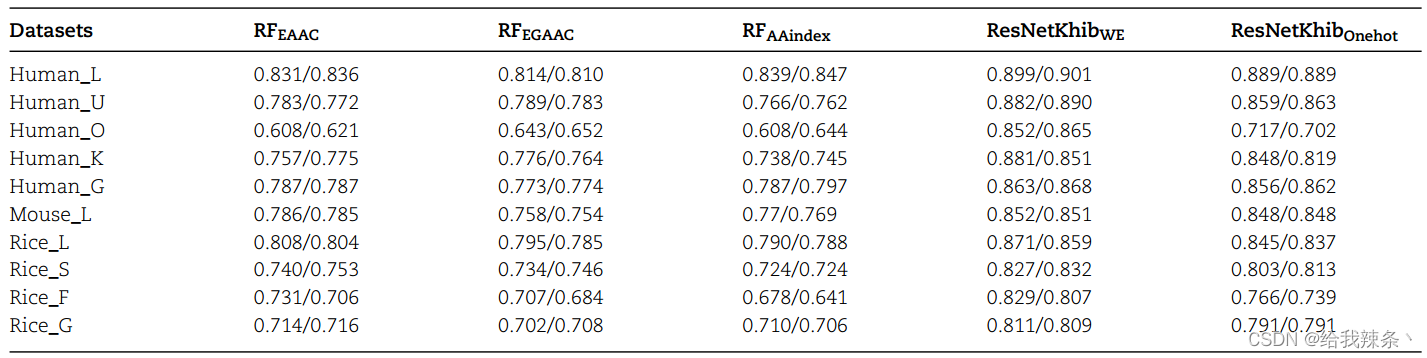
我们可以看到,基于ResNet的模型明显优于基于RF的模型,其中ResNetKhibWE模型的性能最好。对于ResNetKhibWE模型,10个数据集的AUROC值范围为0.807至0.901。然而,基于RF的模型的AUROC值在0.525至0.847之间,表明基于ResNet的模型可以有效地捕捉每种细胞类型的Khib位点的关键特征。
为了可视化ResNetKhibWE模型学习到的特征,我们使用t-SNE[52]从嵌入层、最后一个卷积层和全连接层的输出,可视化了基于独立数据集的样本分布。结果如补充图S4所示。我们可以看到,在包埋层中,阳性和阴性样本混合在一起。然而,这两种类型的样本在卷积层之后明显地彼此分离,并且在完全连接层之后进一步分离。这表明ResNetKhibWE的深度学习框架可以有效地学习特征表示,从而区分正样本和负样本。
不同数据集上预测模型的评估
在这项研究中,我们使用基于ResNetKhibWE框架的每个数据集来训练模型。不同数据集上模型的AUROC值的混淆矩阵如图4所示,其中x轴表示模型,而y轴表示数据集。混淆矩阵中的值表示模型在相应数据集上的性能。
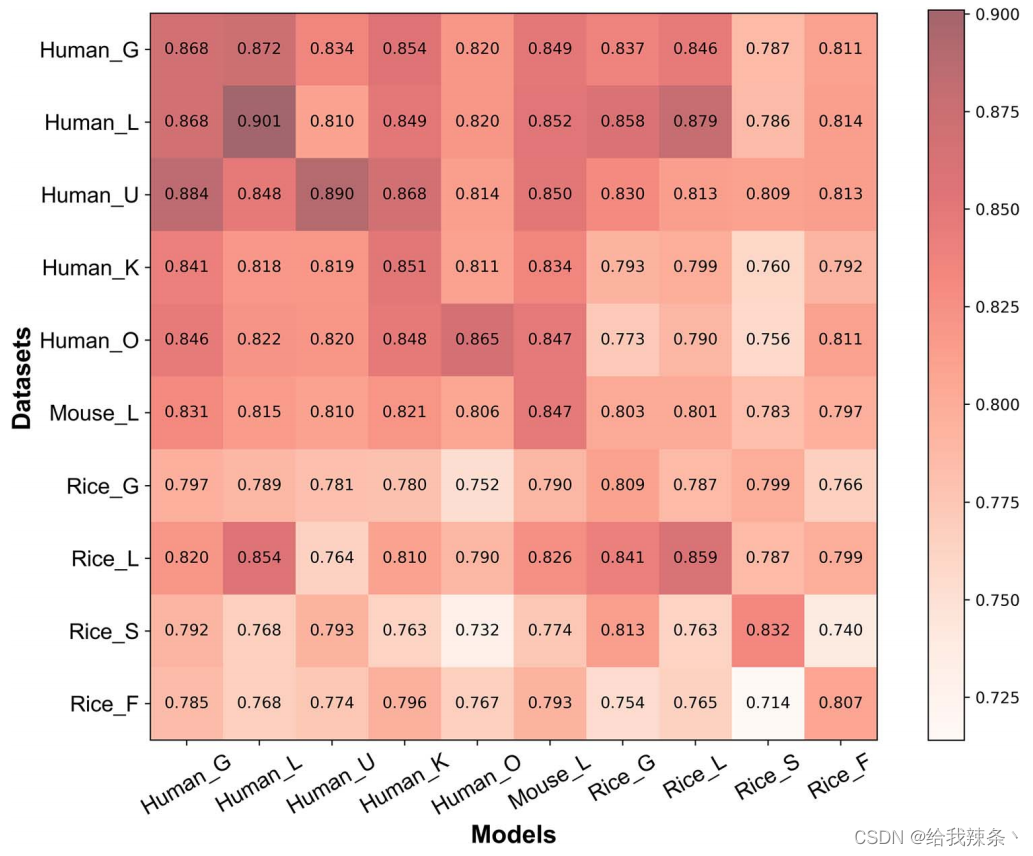
图4. 不同数据集上模型性能的混淆矩阵。x轴表示数据集,而y轴表示模型。矩阵中的值表示相应数据集上模型的AUROC值。
可以看出,对于每个细胞类型特定的数据集,对角线位置具有最高的AUROC值,表明细胞类型特定模型比一般模型表现得更好。例如,人类通用模型在human_L数据集上获得了0.868的AUROC值,而细胞类型特异性模型将AUROC数值进一步提高到0.901。我们还比较了在没有迁移学习策略的情况下训练的细胞类型特异型模型的性能(表3)。
表3. 基于10倍交叉验证和独立测试的模型训练策略的性能比较
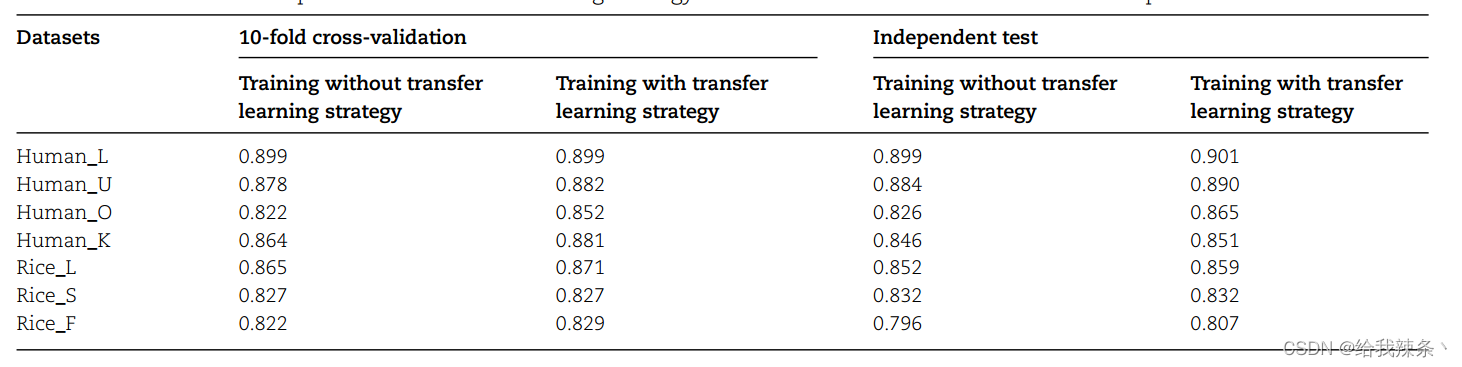
对于大多数细胞类型特定的数据集,迁移学习策略可以提高其预测性能,尤其是对于样本量较小的数据集。以human_O数据集为例,只有669个样本的human-O数据集,这显然不足以训练一个模型。在与人无关的测试数据集上使用迁移学习策略,AUROC值从0.826提高到0.865,这表明迁移学习不仅可以加速训练过程,还可以提高模型的预测性能。
此外,为了进一步评估ResNetKhib的稳健性,我们还重建了具有30%序列同一性的训练数据集,并对模型进行了重新训练。从补充表S11中可以看出,与在具有40%序列同一性的数据集上训练的模型相比,七个再训练模型的性能略有下降,而三个再训练的模型的性能有所提高
与最新预测器相比
我们选择ResNetKhibWE模型作为最终预测模型。为了说明ResNetKhib的预测能力和稳健性,我们在独立测试数据集上进一步比较了ResNetKhib与其他现有Khib位点预测因子的性能
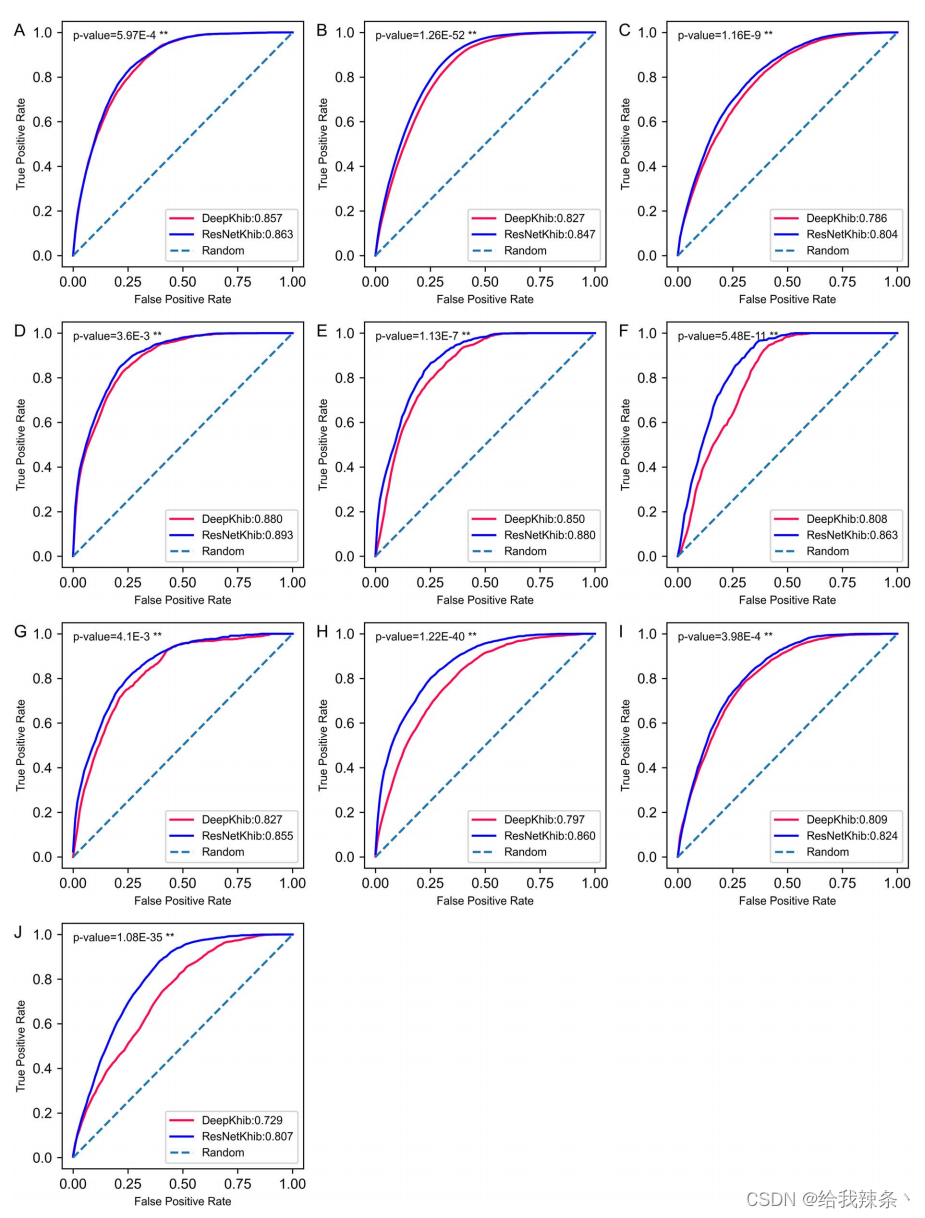
为了进行公平的比较,我们从DeepKhib的训练数据集中使用的独立测试数据集中删除了序列。因此,ResNetKhib在所有数据集上的表现都优于DeepKhib(表4、图5和补充图S5),突出了开发用于Khib位点预测的细胞类型特异性预测因子的重要性和必要性。
表4. ResNetKhib和DeepKhib在五个主要性能指标方面的预测性能,即Acc、Sn、Sp、F1分数、MCC
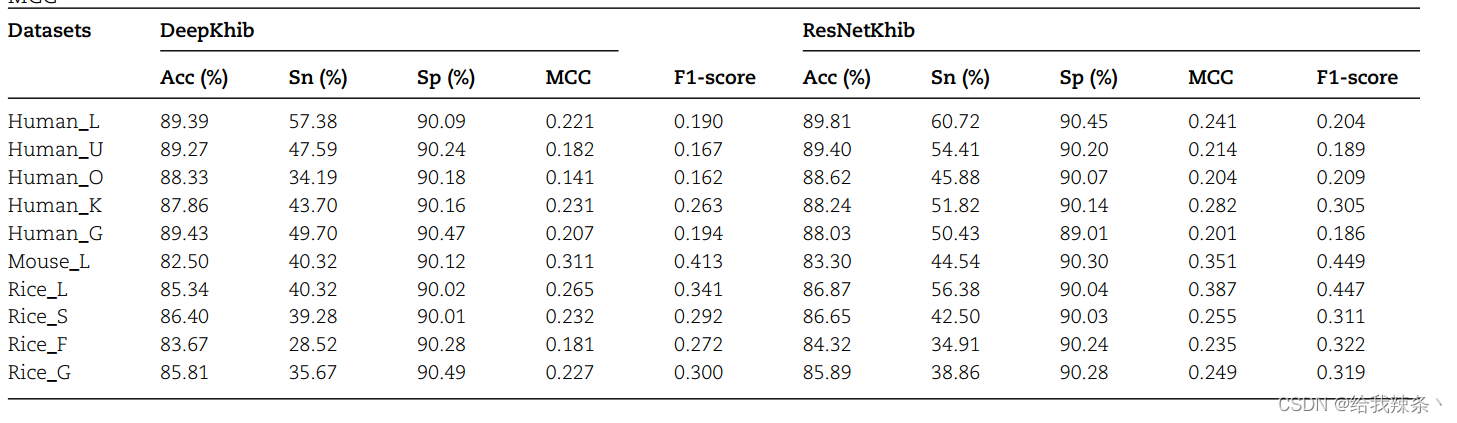
图5. 所提出的ResNetKhib和最先进的预测器DeepKhib在human_G数据集(A)、mouse_L数据集中(B)、rice_G数据集(C)、human-L数据集(D)、human-U数据集(E)、human-O数据集(F)、humar_K数据集(G)、rice-L数据集(H)、rice_S数据集(I)和rice_F数据集(J)的性能比较。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









