





EMDLP: Ensemble multiscale deep learning model for RNA methylation site prediction
期刊:BMC Bioinformatics
论文出处:https://doi.org/10.1186/s12859-022-04756-1
代码链接:https://github.com/whl-cumt/EMDLP
目录
摘要
背景:
最近的研究表明,通过转录后RNA修饰的外转录组调控对所有种类的RNA都是必不可少的。准确识别RNA修饰对于理解它们的作用和调控机制至关重要。然而,传统的RNA修饰位点鉴定实验方法相对复杂、耗时、费力。
机器学习方法已被应用于RNA序列特征提取和分类的计算过程中,可以更有效地补充实验方法。近年来,卷积神经网络(convolutional neural network, CNN)和长短时记忆(long -term memory, LSTM)因其在表示学习方面的强大功能,在修饰位点预测方面取得了很大的成就。然而,CNN可以从空间数据中学习局部响应,但不能学习序列相关性。LSTM专门用于序列建模,可以访问上下文表示,但与CNN相比缺乏空间数据提取。因此,利用自然语言处理(NLP)、深度学习(DL)构建预测框架具有很强的动机。
结果:
他的研究提出了一个集成多尺度深度学习预测器(EMDLP),以NLP和DL的方式识别RNA甲基化位点。它将膨胀卷积和双向LSTM (BiLSTM)有机地结合起来,有助于更好地利用局部和全局信息进行站点预测。
EMDLP的第一步是用NLP的方式表示RNA序列。因此,我们采用了RNA词嵌入编码、One-hot编码和RGloVe三种编码方法,RGloVe是一种基于GloVe的词向量表示改进学习方法,从局部信息和全局信息的角度对站点进行解码。然后,利用扩展卷积神经网络(DCNN)和BiLSTM构建一个扩展卷积双向LSTM网络(DCB)模型,提取潜在的有助于甲基化位点预测的特征。最后,通过软投票将这三种编码方法进行集成,以获得更好的预测性能。
研究背景:
大多数设计用于从序列中进行位点预测的机器学习方法通常首先基于人类理解的特征方法提取特征,然后用分类器预测该位点是否为甲基化位点。例如:
1.RAMPred基于核苷酸化学性质(NCP)、核苷酸组成(NC)提取特征,并首次采用支持向量机(SVM)预测m1A甲基化位点[17]。
2.iRNA-3typeA基于NCP提取特征,累积核苷酸频率(cumulative nucleotide frequency, ANF),采用SVM预测m1A、m6A和A-to-I修饰位点[18]。
3.iMRM基于NCP、NC、One-hot encoding、Dinucleotide Binary encoding (DBE)、Nucleotide Density (ND)、Dinucleotide physicalchemical properties (DPCP)提取特征,采用eXtreme Gradient Boosting(XGboost)对m1A、m6A、m5C、ψ和A-to-I修饰位点进行预测。
众所周知,RNA-seq包含丰富的生物特征信息。因此,RNA序列的理性表达变得更加关键。为了解决这个问题,自然语言处理(NLP)对序列的表示学习(representation learning of sequences of natural language processing, NLP)引起了很大的关注,[25]将RNA序列视为一个句子,k-monomeric unit (k-mer)视为一个单词,得到了很大的支持[26,27]。与传统的机器学习方法相比,大多数深度学习(DL)模型可以分为三个部分:第一,通过NLP模型[28]学习输入数据表示;二是对已学习到的词向量进行合成;第三,通过分类器进行分类,预测该位点是否为甲基化位点。
Gene2Vec[30]、DeepPromise[12]和EDLm6Apred[16]是甲基化位点预测最具代表性和先进的方法。
其中,基于Word2vec[31]和卷积神经网络(convolutional neural network, CNN)开发了Gene2Vec来预测m6A site。
DeepPromise采用CNN和整合增强核酸含量(ENAC)[32]、RNA嵌入词[33]、One-hot编码[20,34]特征识别m1A和m6A位点。
EDLm6Apred采用Word2vec、One-hot编码、RNA词嵌入、BiLSTM等方法预测m6A位点。
然而,现有的方法有以下缺点:
1.从NLP的角度来看,ENAC、One-hot和RNA词嵌入主要关注局部语义信息[16],而忽略了上下文和全局信息。 .Word2vec编码考虑了上下文窗口信息,忽略了全局信息[35]。
2.从DL的角度来看,CNN可以从空间数据[25]中学习局部响应。卷积核的不同尺度会影响网络的学习能力。Gene2Vec[30]和DeepPromise[12]直接使用了由单尺度卷积核组成的CNN,这可能会导致序列[36]的表示学习不完全。两种方法中缺失的信息可能对最终的位点预测很重要。
此外,CNN没有记忆功能,缺乏记忆能力。缺乏[25]学习顺序相关性的能力。相反,EDLm6Apred[16]提出了一个深度的BiLSTM网络来解决上述问题,它可以同时访问上下文信息。但是BiLSTM相比CNN缺乏空间数据提取,训练时间长[37,38]。
考虑上述问题。本文提出EMDLP以NLP和DL的方式识别RNA甲基化位点。其中,One-hot编码、RNA词嵌入和RGloVe是最初编码序列的方法。其次,利用DCNN和BiLSTM构建DCB模型,提取潜在的有助于甲基化位点预测的特征;第三,采用上述三种特征编码方法,在DCB模型的基础上构建了三个预测因子。最后,利用平均预测概率的软投票形成EMDLP,利用三个预测因子获得更好的预测性能。结果表明,在独立测试中,EMDLP模型的性能优于DeepPromise[12]和EDLm6Apred[16]等最先进的方法。
数据和方法
数据集
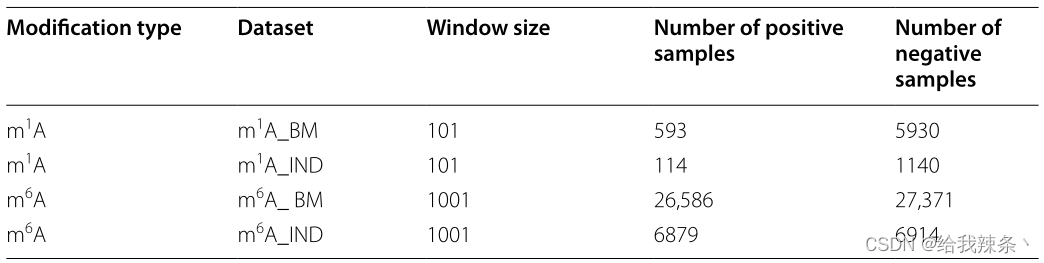
我们提取了两种常见类型的单核苷酸分辨率的人类RNA修饰位点数据集,包括m1A和m6A。对于m1A和m6A站点,本文的数据集分别来源于Chen等人[12]和Zou等人[30]的前期研究。唯一不同的是在m6A站点上使用了Zou验证集作为本文的独立测试集。
该研究将数据集分为两部分:用于交叉验证测试的基准数据集和用于独立测试的独立数据集。以每个样本的改良/未改良位点为中心,取(2n + 1)-nt部分序列窗口。值得注意的是,这两种修改的“n”是不同的。参考Chen论文的实验结果,m1A位点[12]和m6A位点[12]的最佳窗口大小分别为101和1001。如果原始序列的长度小于2n + 1,空的位置将用字符“-”填充,以保证序列长度的一致性。m1A位点和m6A位点阳性样本与阴性样本之比分别为1:10和1:1。这两个RNA修饰数据集的统计结果如表6所示。
表6:这两个RNA修饰数据集的统计
不同角度的特征编码表示
众所周知,特征编码是评价站点预测模型性能好坏的关键。本文采用RNA字嵌入、One-hot编码和RGloVe编码方法对这些序列进行编码。
One-hot编码是一种稀疏的二进制高维词向量,而RNA词嵌入是一种连续的、低维的密集词向量,捕捉局部语义信息。RGloVe继承了GloVe捕获全局语义信息的原理。
One-hot编码是描述核苷酸序列的一种非常简单的编码方法。四个核苷酸和间隙符号“-”被编码为 {A、C、G、T、−},,= (1,0,0,0,0),C = (0, 1, 0, 0, 0), G = (0, 0, 1, 0, 0), T =(0, 0, 0, 0),和“-”=(0,0,0,0,1)。以m1A为例,将一个101nt的序列变换为505位的向量。
RNA词嵌入是RNA序列编码的一种标准方法。大小为k的滑动窗口通过重叠相等长度的RNA序列来滑动,形成k-mer子序列,这些子序列被创建为一个词汇表。以m1A为例。101nt的序列通过大小为3的滑动窗口被转换为99个子序列。研究得到了105个不同的子序列,用唯一整数索引进行索引。每个预处理序列用整数索引改变,并输入Keras嵌入层生成300维的词向量。因此,将101nts序列转化为一个99 × 300的矩阵。
RNA词嵌入只考虑频率信息,而忽略上下文和全局信息。Word2vec只根据每个局部上下文窗口的信息独立训练,而不使用全局共现矩阵[35]中的统计数据。
Pennington等人[40]提出了可以考虑全局共现矩阵中的统计数据的全局向量(GloVe),并使用Adagrad训练手套词嵌入[41]。但是,Adagrad有一个主要的缺点,它会导致Adagrad的学习率下降,变得非常小,此时算法无法学习新的信息[41]。因此,本研究使用RMSProp代替Adagrad来最小化全局矢量模型的损失函数。用这种方法训练出来的词向量称为RGloVe。
k-mer发生率统计是学习嵌入表示的最重要的数据源。Y表示共现计数矩阵,Yij记录了单词k-mer j在单词kmer i的上下文滑动窗口中出现的频率。i, j∈[1,W]是两个k-mer指标,词汇量W = 105。根据GloVe模型,我们通过训练下面的代价函数得到嵌入向量,其中e∈RD是期望的嵌入向量,Qe∈RD是独立的上下文k-mer向量,帮助我们得到e, b, ~b∈R分别是e, ~e的偏差。F (y)是一个非下降的加权函数,其中ymax是一个最大的截止值,β表示分数幂缩放,通常是0.75。
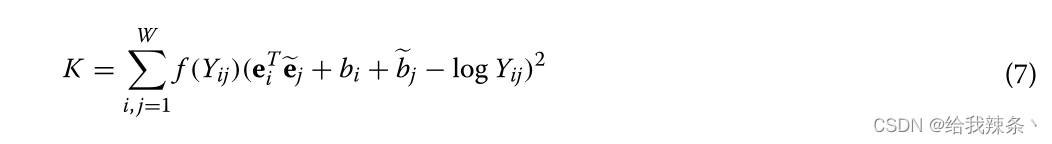

原GloVe使用Adagrad[42]最小化Eq.(7),在每一步t时,具体迭代规则如下:

式中zt, I表示目标函数的梯度,φt, I为时间步长t的参数。每个时间步t对每个参数φt, i的Adagrad更新如下:

其中α表示学习率,Zt, ii∈Rd×d是一个对角矩阵,其中每个对角元素i, i是梯度的平方和。φt, I到时间步长t, δ一般为1 e−8。
Adagrad的主要缺陷是它在分母上累积了平方梯度,此时算法停止学习新信息[41]。RMSprop算法通过降低学习率的单调递减来解决这一缺陷。RMSprop不累积所有过去的平方梯度,但将累积过去梯度的窗口限制为一个固定大小的ξ值。梯度的总和递归地定义为所有过去平方梯度的衰减平均值,而不是仅仅保持ξ以前的平方梯度[41]。在时间步t时,运行平均值E?z2?t取决于之前的平均值E[z2]T−1和电流梯度z2t:

在每一个时间步t,下面每个φt参数的RMSprop更新:

动量项(入)通常设置为0.9或类似的值,而RMSprop α的学习率为0.001。我们使用RMSprop最小化Eq.(7),得到d维嵌入向量表示e1, e2, e3,…eW∈RD。根据向量,本研究通过将每个k-mer嵌入向量空间RD,完成表征学习fembedding(x): CL→RL×D的嵌入编码:

式中x = [x1, x2, x3,…]∈CL, xL]我们根据输出的L × D矩阵进行了卷积阶段。
以m1A为例。如果维度为300,则将101nts序列转换为99 × 300的矩阵。表7列出了三种特征编码输入和输出格式。
表7:输入输出格式采用三种特征编码

当应用于一维情况时,扩张卷积的计算公式为式(14)。

其中xj为输入的第j个元素,yj为DCNN中第j个元素的输出,ω为滤波器的权值,N为滤波器的长度,r为DR。
除了膨胀的卷积,DCNN还包括pooling和dropout层。池化层应用于每个feature map,并在池化窗口中输出输入的平均值或最大值,这样池化层可以减少参数的数量。
dropout层用于避免模型训练时的过拟合,是最常用的正则化技术。在转发传播过程中的每次训练活动中,都会随机将一些神经元设为零,这直观地导致了不同网络的融合。退出率是神经元退出的概率。
在本研究中,我们将三种扩张速率(DR = 1、2、3)的扩张卷积层串联起来送到BiLSTM阶段。
基于扩张卷积双向LSTM的站点预测
本研究将DCB模型与RNA词嵌入、一热编码和RGloVe三种编码方法相结合,构建了三种修饰位点预测因子。考虑RGloVe预测器,如图7所示。
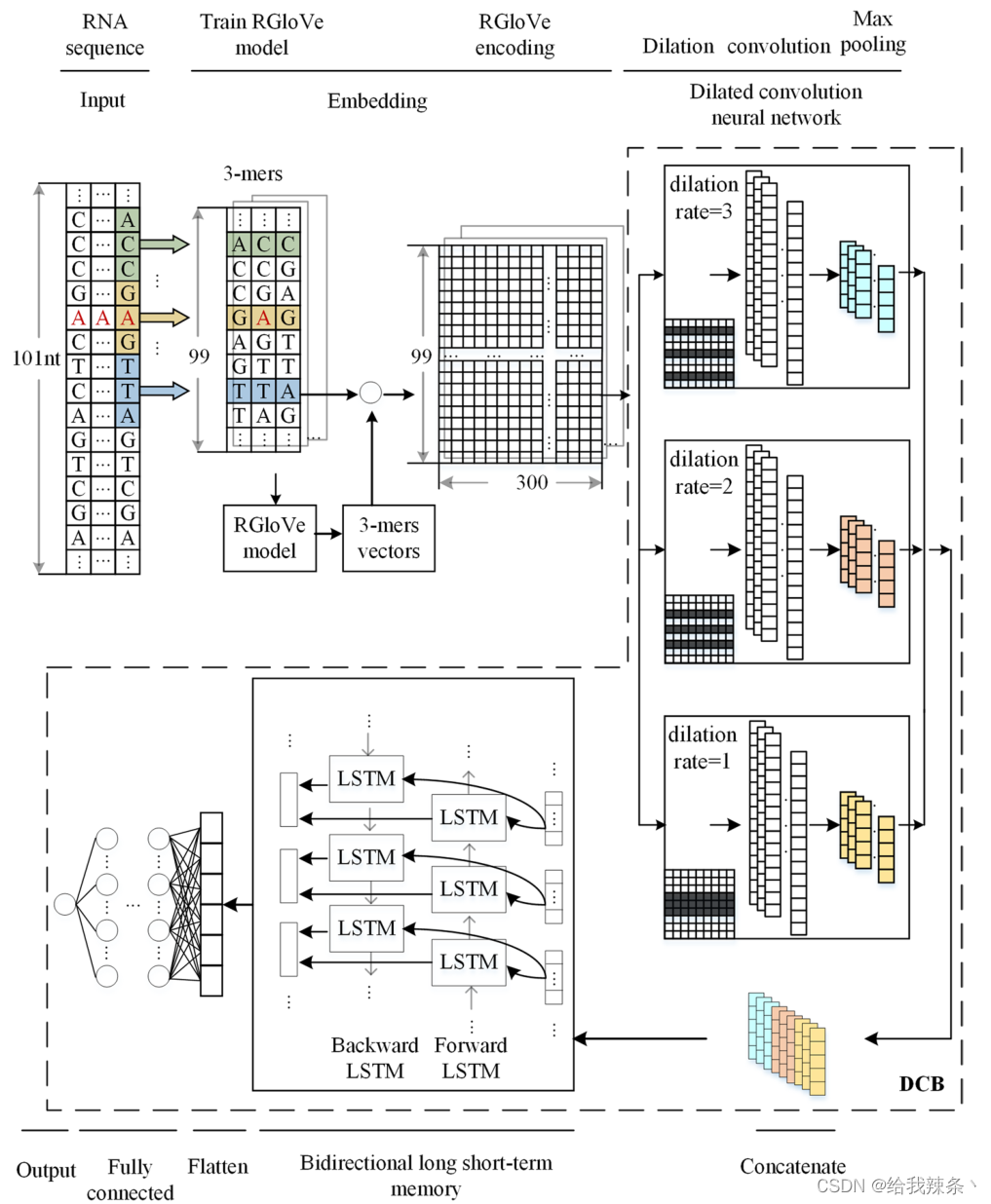
图7:基于RGloVe、DCNN和BiLSTM神经网络的计算框架结构来预测m1A的甲基化位点
假设我们有N个长度为10的RNA序列。每个样品都有一个二值标签,表明是否为甲基化修饰位点,即n标记的样品{xn, yn}Nn=1 yn∈{0,1}。对于每一个包含A、C、T、G核苷酸和“-”的序列xn,我们通过一个拆分窗口将其拆分为子序列。每个包含k个核苷酸的子序列被称为k-mer基序。我们提取步幅为s的长度为k的子序列,得到长度为L = [(L0−k)/s] + 1的k-mer motif。以m1A为例。将L0 = 101nts的序列通过大小k = 3、步幅s = 1的拆分窗口转换为99个子序列,其中所有这些3-mers在集合C =[1,2,3,4…,105]和序列数据x∈CL中都具有正整数指标。
下面的内容将专门介绍学习一个特征映射f: CL→Rd,它将x∈CL映射到对DL任务有用的特征向量h∈Rd。
我们使用带有k-mer嵌入的DCB来训练模型,如图7所示。表示学习函数f: CL→Rd可以分为四个阶段:

嵌入阶段计算k-mers的共现统计量,并将它们映射到d维空间RD。
DCNN阶段有3块DCNNs, 3块DCNNs的稀释率分别为1、2、3。每个DCNN块都包含一个以整流线性单元(ReLU)为活动函数的扩张卷积层、一个max-pooling层和一个dropout单元。我们采用网格搜索策略对超参数进行优化。有64个卷积核,每个核的大小为3。对于max-pool层,max-pool窗口的大小是2。drop rate设置为0.2,避免过拟合。串联阶段将三个dcnn块串联起来,构建一个多尺度特征提取器。BiLSTM阶段对输入应用双向LSTM网络,以便收集数据之间的长期数据依赖信息。
神经元数量设为64个,下降速率为0.2。BiLSTM阶段结束后,数据由平坦层平展成一维,然后是全连通层。全连接层由3个全连接组成,其中包含的神经元数量为256,128,64个,由ReLU函数激活,dropout概率为0.5。最后,输出层计算概率得分,表示该站点被Sigmoid函数修改的可能性如下:

位点预测
不同的编码技术将从不同的角度观察序列。RNA词嵌入和One-hot编码强调局部信息,而RGlove利用全局统计来学习全局语义。因此,不同的预测因子对预测的影响可能是互补的。在DCB模型的基础上,通过RNA词嵌入、One-hot编码和RGloVe构建了三个预测因子。最后,利用上述三个预测因子通过软投票形成EMDLP,如图8所示。
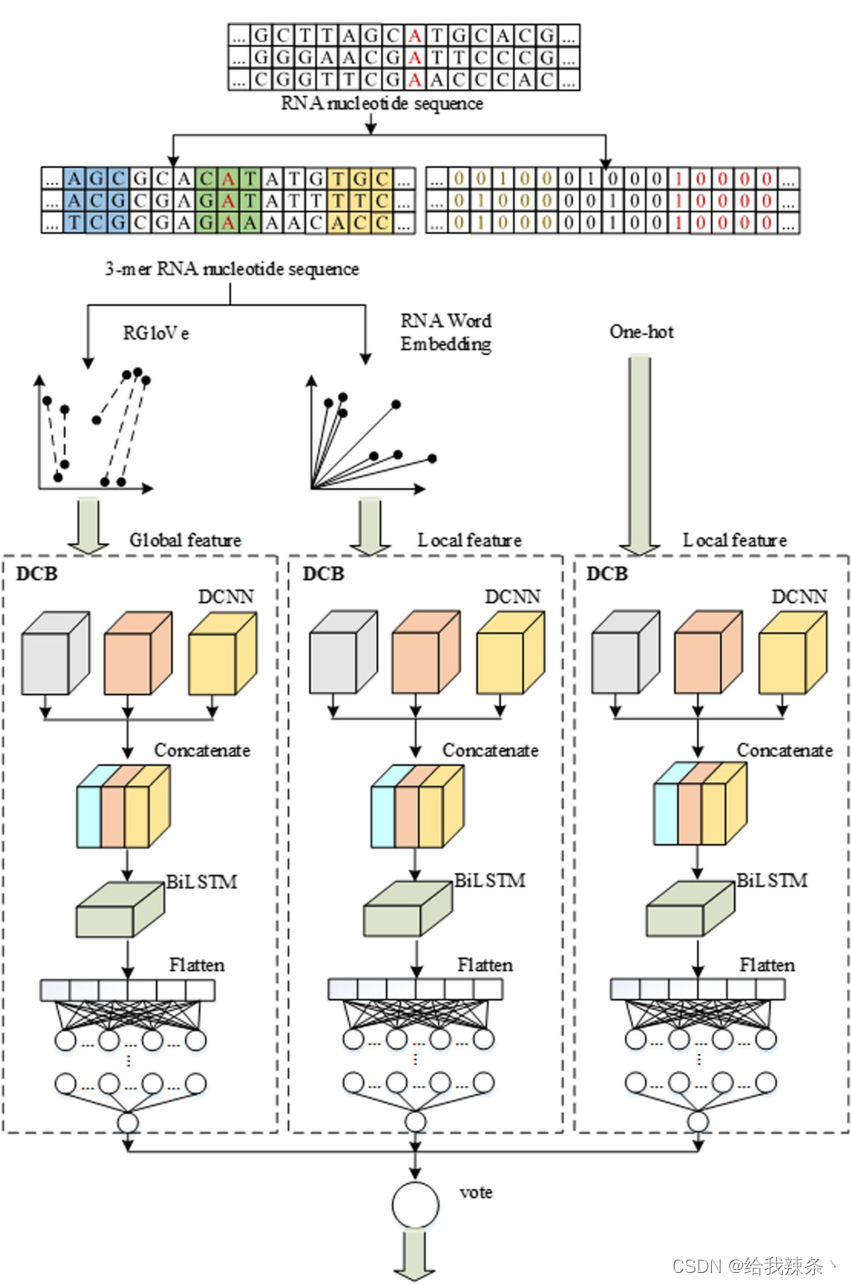
图8:EMDLP预测器的结构
研究结果:
评估指标
为了估计模型的预测,我们采用了广泛使用的二元分类器评价指标,包括Sensitivity(Sn, Recall)、Specificity(Sp)、Accuracy(Acc)、Precision(Pre)、F1 score (F1)、Matthews correlation coefficient(MCC)、Area under the receiver operating characteristic(AUROC)和Area under the Precision - Recall curve (AUPRC)。Sn、Sp、Acc、Pre、F1、MCC的定义如下:

其中TP为真阳性,TN为真阴性,FP为假阳性,FN为假阴性。此外,根据受试者操作曲线(ROC)和精确召回曲线(PRC)分别计算AUROC和AUPRC值。除MCC取值范围为[−1,+ 1]外,其他指标取值范围为0 ~ 1,数值越大表示性能越好。
结果分析
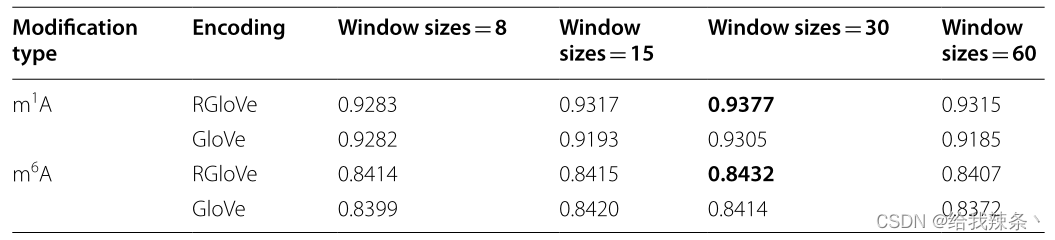
滑动窗口的大小是影响编码方案性能的一个重要参数。基于基准数据集,本实验比较了RGloVe和GloVe在四种不同滑动窗口大小(即:、8、15、30和60)。RGloVe基于GloVe模型框架,采用RMSProp代替Adagrad来最小化全局矢量模型的损失函数。因此,当滑动窗口长度= 30时,RGloVe的预测性能最好,如表1所示。实验结果表明,使用RMSProp可以更有效地训练模型。
表1:基于基准数据集的RGloVe和GloVe在不同滑动窗口大小下的AUROC评分
与其他不同学习模式的比较
接下来,使用相同的基准数据集,将DCB与CNN、DCNN和BiLSTM进行比较分析。实验采用RGloVe编码来描述RNA序列,分别构建了CNNRGloVe、DCNNRGloVe、BiLSTMRGloVe和DCBRGloVe。其中,CNNRGloVe采用了Deeppromise[12]中的CNN模型。DCBRGloVe是一个自建的DCB模型,包括DCNN和BiLSTM阶段。DCNNRGloVe表示去除BiLSTM阶段的DCBRGloVe,该阶段被flatten layer取代。同样,BiLSTMRGloVe代表不带DCNN阶段的DCBRGloVe。
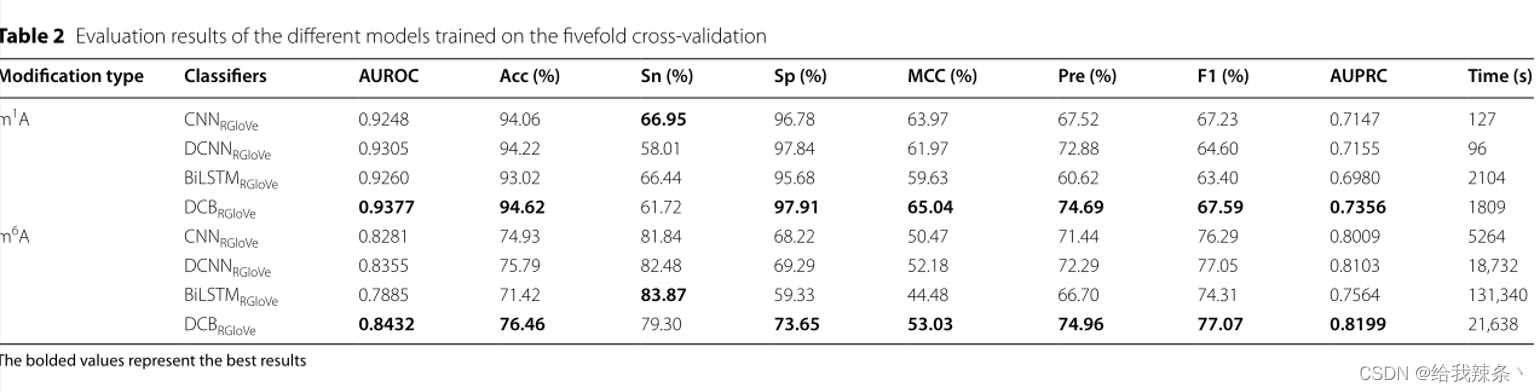
5次交叉验证评价结果,m1A和m6A的AUROC和AUPRC曲线如图2和表2所示。结果显示,DCNNRGloVe在m1A和m6A上的AUROC分别比CNNRGloVe高0.57%和0.74%,AUPRC分别比CNNRGloVe高0.08%和0.94%。
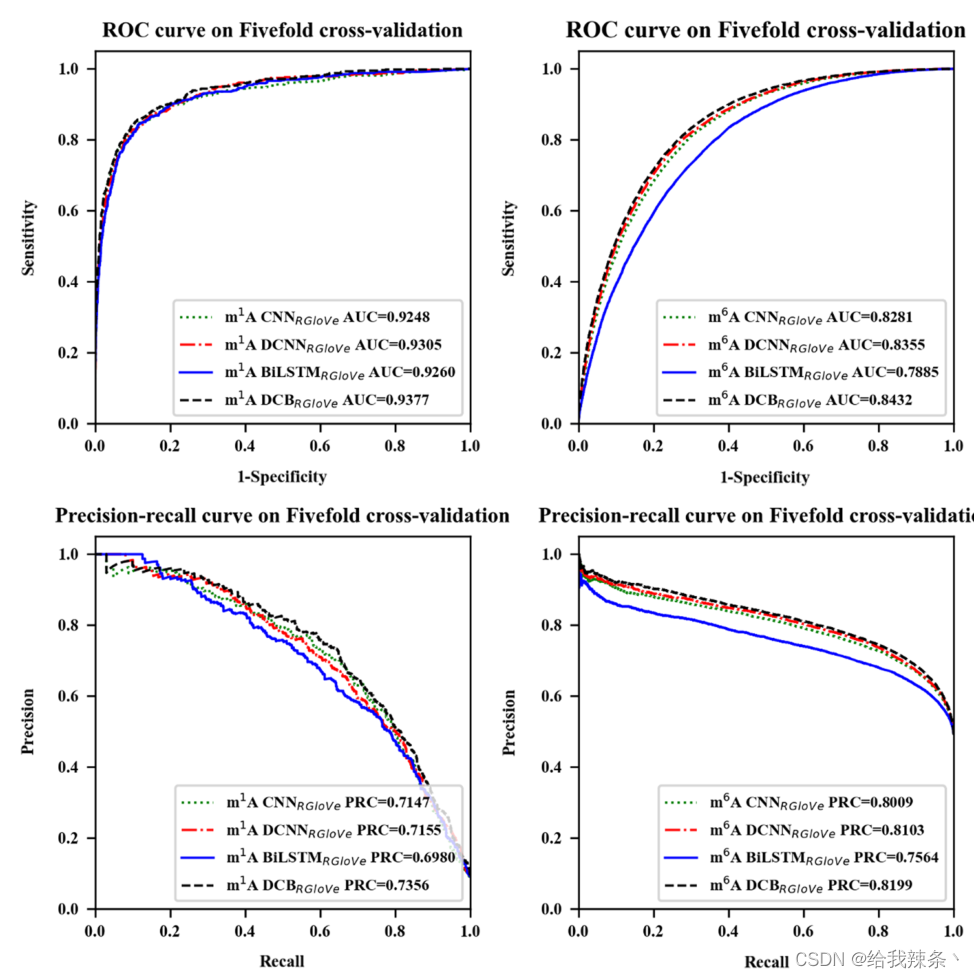
图2:通过五次交叉验证不同模型的性能。模型分别为CNNRGloVe、DCNNRGloVe、BiLSTMRGloVe和DCBRGloVe。“CNNRGloVe”采用了Deeppromise中的CNN模型;DCBRGloVe代表一个自建的DCB模型,包括DCNN和BiLSTM阶段;“DCNNRGloVe”表示去除BiLSTM期的DCBRGloVe;“BiLSTMRGloVe”表示不带DCNN阶段的DCBRGloVe。
表2:不同模型的评价结果经过五次交叉验证
这个结果验证了CNN中的单尺度卷积核对从RNA序列学习深度语义具有挑战性。相反,多尺度卷积核可以提取额外的特征来提供深度语义。
此外,本研究还比较了DCBRGloVe和DCNNRGloVe的性能。在m1A和m6A上,DCBRGloVe的AUROC分别比DCNNRGloVe高0.72%和0.77%,在m1A和m6A上,DCBRGloVe的AUPRC分别比DCNNRGloVe高2.01%和0.96%。原因可能是DCNN没有记忆功能,不能学习顺序相关性。相反,DCB可以根据DCNN捕捉不同空间结构的局部相关性,并根据BiLSTM有效地学习文本中每个k-mer的上下文。综上所述,DCB方法比其他方法更能准确地理解序列语义。
最后,本研究比较了DCBRGloVe和BiLSTMRGloVe的运行时间。虽然影响模型训练时间的因素很多,但实验结果表明,BiLSTMRGloVe的训练时间很长,是DCBRGloVe的几倍。这是因为DCNN阶段的最大池化层减少了网络的参数,对降低维数和计算复杂度起到了积极的作用。
与其他不同特征编码方法的比较
另外,下面的内容比较了四种特征编码方法的预测性能。实验采用RGloVe和RNA词嵌入、One-hot编码、word2vec三种常用的编码方案分别对序列进行编码,然后应用相同的DCB模型在相同的独立数据集上预测修改位点。对比结果表明,RGloVe对AUROC的预测优于其他三种编码技术,如图3和表3所示。在准确意义上,对于m1A和m6A位点,DCBRGloVe的AUROC分别为0.9468和0.8486,比其他方法更准确。原因在于One-hot编码和RNA词嵌入强调局部语义信息,Word2vec编码强调上下文窗口信息,而上述三种编码都忽略了全局信息。RGloVe继承了GloVe的优点,它结合了全局矩阵分解和局部上下文方法[37]的优点。因此,RGloVe可以利用这一优势来提高模型的预测精度。总之,RGloVe比其他三种常用方案具有更高的语义精度。
表3:基于One-hot编码、RNA词嵌入、Word2vec和RGloVe的DCB模型评价结果
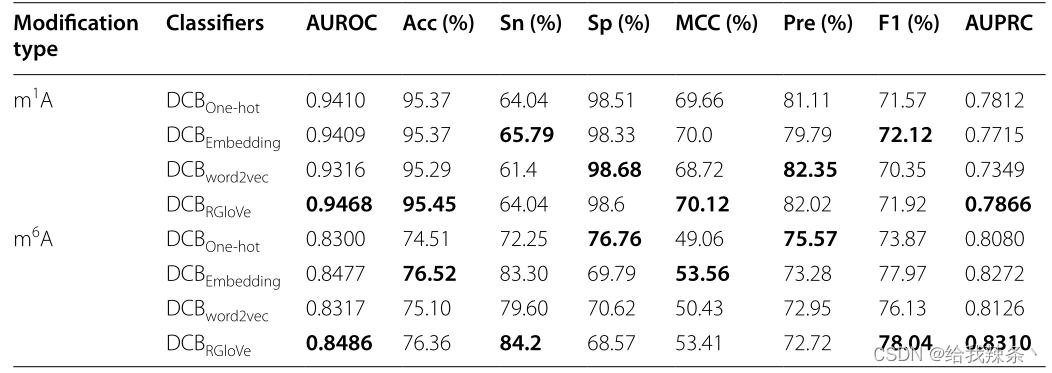
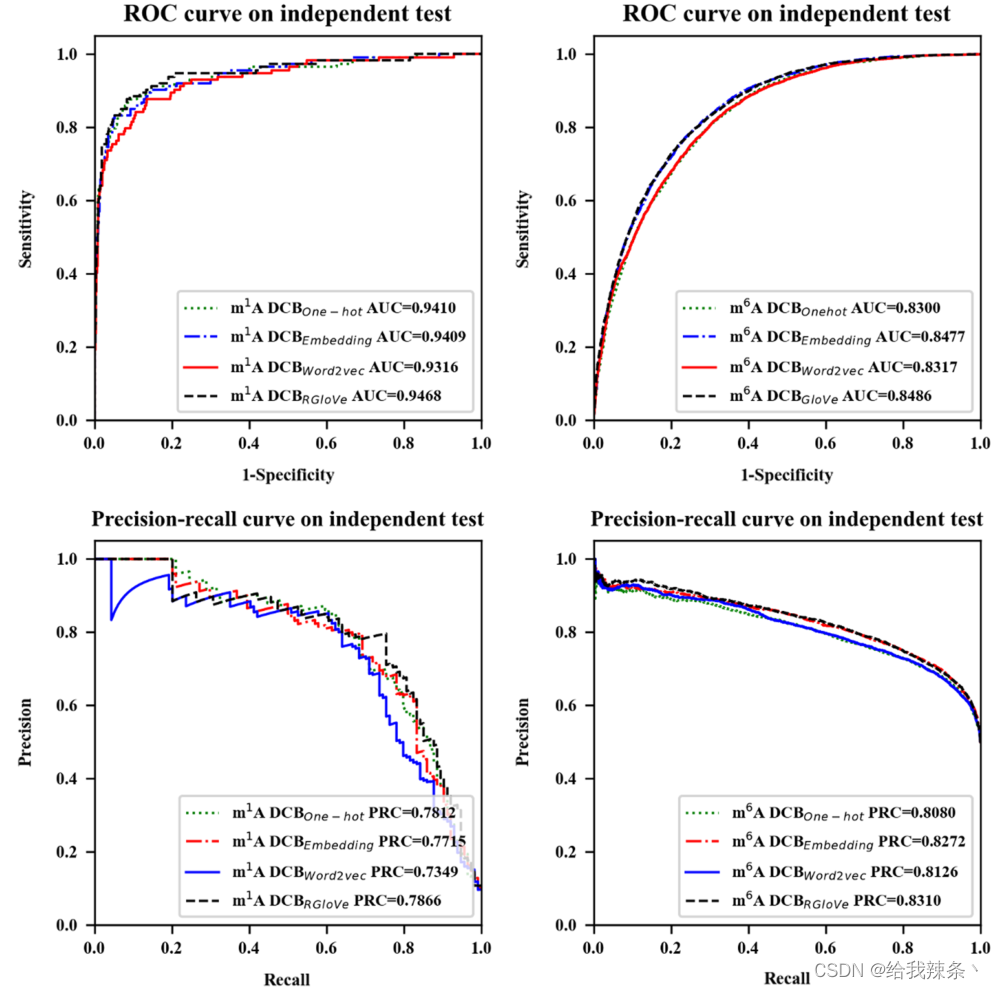
图3:基于One-hot编码、RNA词嵌入、Word2vec和RGloVe的DCB模型的性能
与最先进的方法进行比较
最后,在相同的独立数据集上,将EMDLP与其他最先进的方法进行了比较,如DeepPromise[12]和EDLm6Apred[16]。为了使比较更加说明问题,我们用DCB替换了DCBDeepPromise中的CNN模型来构建DCBDeepPromise,我们的EMDLP用RGloVe替换了DCBDeepPromise中的ENAC编码。
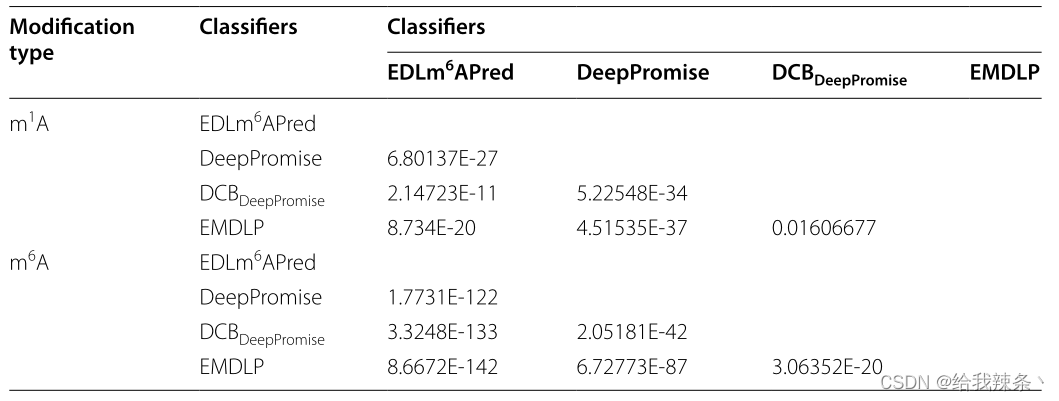
为了评估模型的可靠性,在相同的m1A和m6A独立测试集上分别对EDLm6Apred、DeepPromise、DCBDeepPromise和EMDLP模型进行了100次重复实验。在每个重复中,产生新的评价结果。如图4、表4、图5所示,EMDLP的AUROC和AUPRC优于其他方法。原因可能是ENAC、One-hot和RNA的词嵌入主要关注局部语义信息,Word2vec编码考虑上下文窗口信息,而不关注全局统计信息。同时,RGloVe可以比其他四种编码更全面地表示语义信息序列。与其他方法相比,DCB更适合于提取RNA序列的特征。此外,我们使用学生t检验[39]检验不同工具间AUROC值的统计显著性,如表5所示。
表4:比较EMDLP模型
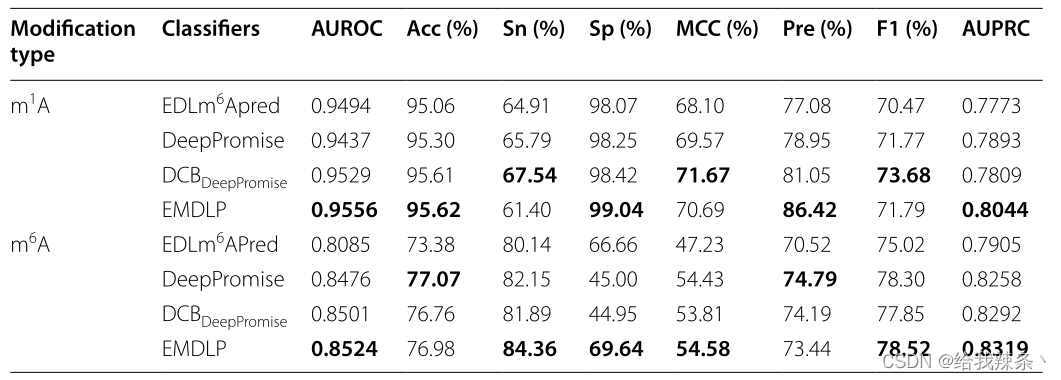
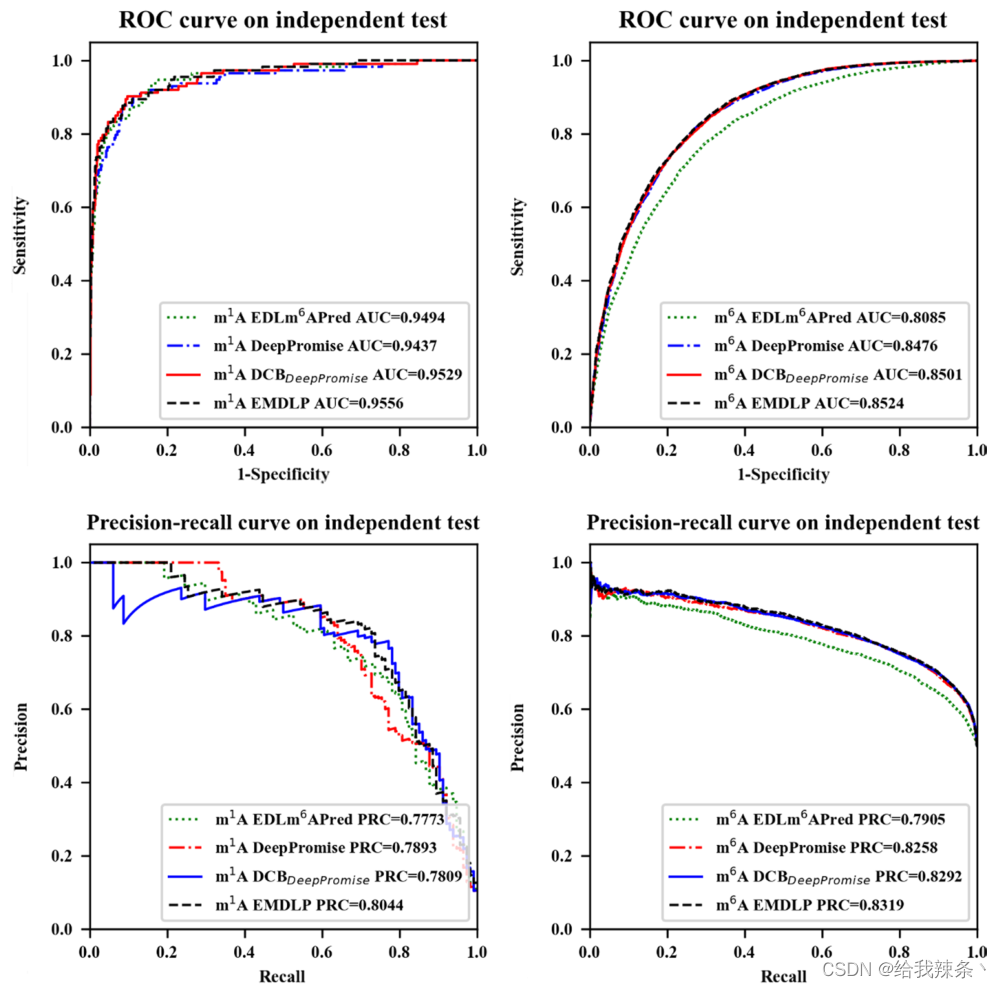 图4:EMDLP和其他方法在独立测试中的表现
图4:EMDLP和其他方法在独立测试中的表现
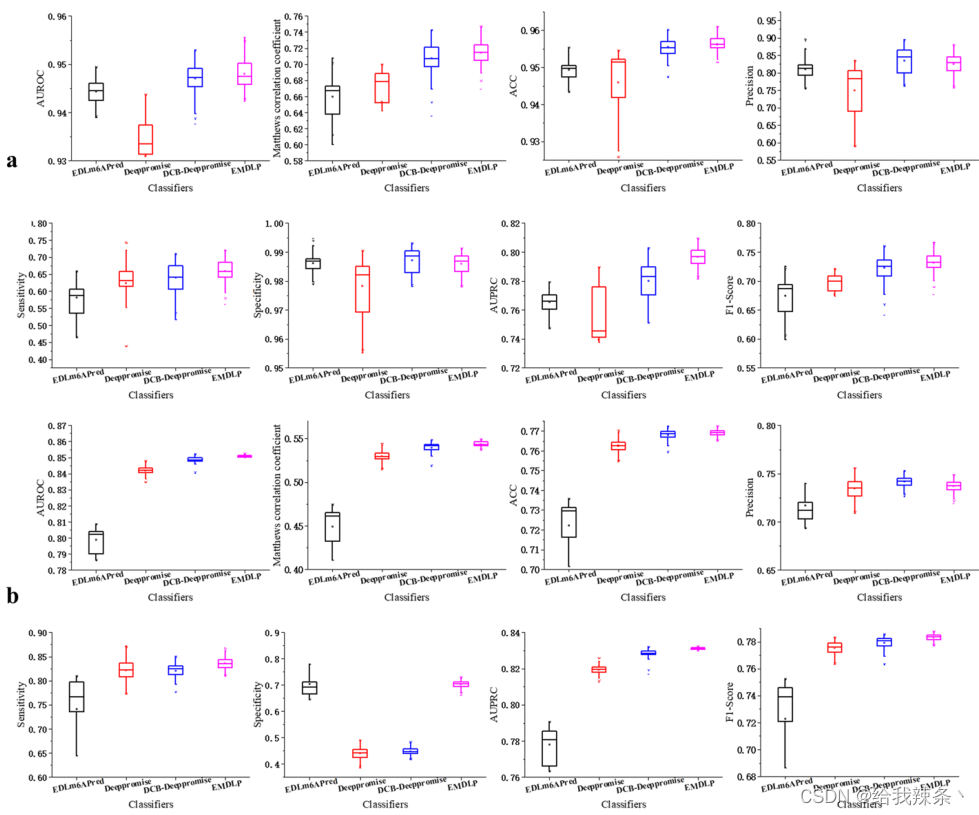
图5:基于四种方法100次重复的paperformance对四种方法的比较性能评估的8个指标的箱线图。a表示m1A独立的数据集。b表示m6A独立数据集
表5:四种分类器性能的差异有统计学意义的相关矩阵
讨论
本文提出EMDLP以NLP和DL的方式识别RNA甲基化位点。具体讨论如下:
首先,本研究比较了基于RGloVe和GloVe编码方法在4种不同滑动窗口大小(8、15、30和60)下对m1A和m6A甲基化位点的预测性能。评估结果表明,使用RMSProp代替Adagrad来最小化全局向量模型的损失函数,确实可以更有效地训练模型。这一结果与Ruder, S.(2017)的结果一致,Ruder, S.(2017)指出RMSProp可以克服Adagrad的弱点。当滑动窗口长度= 30时,RGloVe的预测性能最好。
其次,基于上述RGloVe对序列的特征表示,本研究将DCB模型与预测甲基化修饰位点的CNN、DCNN和BiLSTM模型进行了比较。实验结果表明,DCNNRGloVe在m1A和m6A上的AUROC分别比CNNRGloVe高0.57%和0.74%。本研究证实了多尺度卷积核可以提取不同的特征来提供深度语义。实验结果表明,BiLSTMRGloVe的训练时间很长,是DCBRGloVe的几倍。这也符合Min, x的结论,DCNN阶段的max-pooling层减少了网络的参数,对降低维数和计算复杂度起到了积极的作用。实验结果表明,DCBRGloVe模型在预测m1A和m6A位点方面优于其他模型。本研究证实了DCNN与BiLSTM的结合使得序列语义的理解更加准确。
第三,基于上述自建的DCB模型,比较了RGloVe、RNA词嵌入、One-hot编码和word2vec的预测性能。结果表明,我们的RGloVe在预测性能上优于其他三种编码方案。这一发现与Pennington, J(2014)提出的GloVe比word2vec具有更高的语义准确率是一致的。
最后,通过软投票构造EMDLP,使用三个预测因子获得更好的预测性能。本文比较了基于独立数据集的EMDLP、DeepPromise、DCBDeepPromise和EDLm6Apred的预测性能。结果表明,EMDLP的AUROC明显优于三种方法。本研究进一步表明,RGloVe比其他四种编码方法更能代表序列的语义信息,而DCB比其他方法更适合提取RNA序列的特征。
结论
本文提出了一种预测因子EMDLP,用于NLP和DL方法识别RNA甲基化位点。它将膨胀卷积和BiLSTM有机地结合在一起,有助于更好地利用局部和全局信息进行站点预测。
尽管EMDLP的性能优于最先进的预测方法,但该方法目前仅限于人类,由于缺乏用于其他物种的足够数量的单核苷酸数据集,尚未扩展到其他模式生物。当未来有足够的其他物种RNA修饰数据集时,对EMDLP的性能进行测试是值得期待的。















 915
915











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









