






PHIAF:通过基于GAN的数据增强和基于序列的特征融合来预测噬菌体-宿主之间的相互作用
摘要
噬菌体治疗已成为抗生素治疗细菌性疾病的最有前途的替代品之一,识别噬菌体-宿主相互作用(PHIs)有助于了解噬菌体感染细菌的可能机制,以指导噬菌体治疗的发展。与湿法实验相比,识别PHIs的计算方法可以降低成本,节省时间,更有效和经济。在本文中,我们提出了一种基于生成对抗网络(GAN)的数据增强和基于序列的特征融合(PHIAF)的PHI预测方法。首先,PHIAF应用基于gan的数据增强模块,生成伪PHIs来缓解数据稀缺。其次,PHIAF融合了来自DNA和蛋白质序列的特征,以获得更好的性能。第三,PHIAF利用一种注意机制来考虑DNA/蛋白质序列衍生特征的不同贡献,这也提供了预测模型的可解释性。在计算实验中,通过5倍交叉验证(AUC和AUPR分别为0.88和0.86),AUC和AUPR的性能优于其他最先进的PHI预测方法。消融研究表明,数据增强、特征融合和注意机制都有利于提高PHIAF的预测性能。此外,在该案例研究中,PHIAF得分最高的4个新的PHIs被最近的文献证实。总之,PHIAF是一种很有前途的加速探索噬菌体治疗的工具
介绍
现有报告表明,细菌感染可能参与各种类型疾病的生长和发展,包括霍乱[、inf炎症性肠病、结肠癌、破伤风和不同类型的癌症。研究人员在1928年发现了抗生素,并在临床实践中使用它们,用于治疗严重的细菌性疾病,挽救了无数的生命。不幸的是,由于抗生素的过度使用,细菌发展了一些耐药机制。2019年,美国疾病控制和预防中心报告称,美国每年约发生280万例耐药感染,导致超过35 000人死亡;在欧洲,大约每年有3.3万人死于耐抗生素感染,。因此,迫切需要开发新的抗生素或替代疗法,以避免抗生素耐药性感染的进一步恶化。然而,由于制药公司生产成本高、预期效益不理想、等原因,许多制药公司不再开发新的抗生素,研发时间长。因此,研究人员希望寻找替代疗法来减少耐抗生素感染和治疗细菌性疾病。
噬菌体不仅可以摧毁特定的细菌宿主,而且还可以以指数级复制,这些特性使噬菌体成为治疗细菌性疾病和治疗耐药感染的最有前途的治疗方法之一。确定噬菌体-宿主的相互作用(PHIs)有助于了解噬菌体是否可以用来治疗细菌性疾病。然而,phi的实验验证需要相当多的时间、人力和金钱。因此,研究人员试图开发计算PHI预测方法,筛选出治疗细菌性疾病的靶噬菌体,并指导体内验证,从而大大减少了所需的时间和成本。
分子和生态共同进化过程塑造了噬菌体和细菌基因组,并在其基因组序列中留下信号,使研究人员能够预测PHI,因此,基于噬菌体和宿主基因组序列的各种PHI计算方法已经开发出。例如,Ahlgren等人。提出了基于DNA序列的病毒主机匹配器(VHM),通过计算噬菌体和宿主的寡核苷酸频率模式之间的距离来预测PHIs。然而,VHM的运行时间阻碍了它在大数据集上的发展,因此Galiez等人提出VHM通过构建马尔可夫模型来预测噬菌体的原核宿主来减少运行时间。与VHM相比,WIsH的运行时间减少了几百倍。除了通过计算噬菌体和宿主之间的相似性来预测PHIs的VHM和WIsH外,研究人员还使用了各种机器学习分类器,包括逻辑回归(LR)、支持向量机(SVM)、随机森林(RF)和朴素贝叶斯(NB)来预测PHIs 。此外,我们还开发了PHP 和VIDHOP 来增强PHI的预测
一些研究表明,蛋白质在噬菌体和宿主的生物过程中起着重要作用;因此,研究人员提出了基于蛋白质序列的PHI预测方法。例如,Leite等人利用噬菌体和宿主蛋白的一级结构序列和经典分类器,包括RF、SVM、LR、k-最近邻(KNN)、多层感知器(MLP)和NB,来预测PHIs。在上述方法的基础上,Li等人使用了卷积神经网络(CNN)来提高PHI预测的性能。
虽然现有的方法在PHI预测方面取得了良好的性能,但仍存在一些挑战。首先,在数据库数据库中有数千个经过实验验证的PHIs,但只有几百个非冗余的PHIs可用,可以用于构建预测模型。这一局限性阻碍了高性能预测模型的开发。其次,现有的方法大多使用噬菌体和宿主的DNA序列或蛋白质序列来构建预测模型,但很少结合两种类型的序列。第三,虽然已经使用了多种特征和机器学习技术来建立预测模型,但这些模型往往缺乏足够的可解释性,这阻碍了对PHIs机制的阐述。
近年来,深度学习技术在生物信息学领域受到了广泛的关注,研究人员将这些技术应用于处理不同的任务。生成对抗网络(GAN)作为深度学习技术的一个分支,最初用于图像处理,后来在数据增强方面表现出良好的性能。例如,Wan等人成功地利用GAN生成了基于蛋白质序列的生物物理特征。同时,研究人员开发了一种用于深度学习的注意机制,以提高预测模型的可解释性,以提高预测性能。这些深度学习技术的发展促使我们进一步加强和改进PHI预测。
在本研究中,我们提出了一种基于GAN数据增强和基于序列的特征融合的新型PHI预测方法,简称PHIAF,以解决PHI预测的各种挑战。首先,PHIAF利用GAN构建了一个数据增强模块,生成高质量的伪样本,以克服PHI数据稀缺的瓶颈。其次,PHIAF融合了噬菌体和宿主的DNA和蛋白质序列编码的不同特征,以提高预测性能。第三,PHIAF利用CNN构建了一个PHI预测模块,并将一个注意机制整合到CNN中,以提供预测模型的可解释性实验结果表明,PHIAF方法优于目前最先进的PHI预测方法。烧蚀研究和讨论表明,CNN中由数据增强模块生成的伪样本、DNA和蛋白质序列衍生特征的融合以及PHIAF的注意机制都有效地提高了PHIAF的性能。
材料和方法
数据
2021年3月,我们从四个广泛使用的数据库中下载数据(包括噬菌体、宿主及其相互作用),其中包括MVP ,PhagesDB ,VHDB 和NCBI ,并将这些数据合并,构建一个具有更多PHIs的数据集,用于我们的研究。对这四个数据库中的数据进行以下处理。首先,我们删除未发表在文献中或未包含在NCBI记录中的PHIs,以确保PHIs是可靠的。其次,我们根据噬菌体的定义删除了错误标记为噬菌体/宿主的数据(噬菌体是在细菌和古细菌中感染和复制的病毒)。移除的数据包括不属于病毒的噬菌体和不属于细菌或古细菌的宿主。第三,我们从NCBI数据库中提取剩余噬菌体和宿主的全基因组序列和编码蛋白序列。
经过上述过程,我们将4个数据库中剩余的噬菌体和宿主结合起来,去除重复的噬菌体,得到5331个噬菌体与235个宿主共发生5399个相互作用。噬菌体的数量远远大于宿主的数量,一个宿主可能与多个噬菌体相互作用。我们使用算法1去除每个宿主具有高相似度的冗余噬菌体(不同相似度度量的比较和噬菌体Pk[0]对预测性能的影响分别见补充材料第1和2节)。我们将0.90设置为一个高相似度阈值,这与CD-HIT工具[41]的默认阈值相同。在减少冗余后,我们获得了一个包含304个噬菌体与235个宿主之间的312个相互作用的基准数据集,可用于更好地评价预测模型的性能。在这个数据集中,我们将312个已知的PHIs设置为阳性样本,并从所有未知的PHIs中选择阴性样本,同时确保阳性样本和阴性样本的数量相等。

PHIAF
PHIAF主要由特征提取、数据增强和PHI预测三个模块组成。PHIAF的示意图如图1所示。首先,噬菌体的DNA和蛋白质序列和主机被编码到特性中(图1A)。其次,使用基于GAN的数据增强模块生成伪PHIs(图1B)。最后,在CNN框架下构建一个PHI预测模块,注意利用DNA和蛋白质序列重塑成适当形式后的特征来预测PHI(图1C)。
特征提取模块
一些研究表明,DNA和蛋白质序列在噬菌体和宿主的生物进化中起着重要作用。为了获得更全面、有效的信息,我们从噬菌体和宿主的DNA和蛋白质序列中提取特征,如表1所示;这些特征的详细信息见补充材料的第3节。
由于噬菌体和宿主的DNA序列具有不同的长度(噬菌体和宿主的DNA和蛋白质序列长度的分布分析见补充材料第4节),我们考虑了一些与序列长度无关的DNA序列衍生特征,包括Kmer、反向互补Kmer(RCKmer)、核酸组成(NAC)、二核苷酸组成(DNC)、三核苷酸组成(TNC)、k间隔核酸对组成(CKSNAP)和三核苷酸的电子离子相互作用假电位(PseEIIP)。我们使用iLearn软件计算了这些特征,并获得了每个噬菌体/宿主DNA序列的340维特征向量。
我们遵循之前的研究,提取了广泛使用的蛋白质序列衍生特征,包括氨基酸组成(AAC)、组成蛋白质的化学元素(AC)的丰度和蛋白质的分子量(MW)。由于每个噬菌体/宿主都有多个蛋白质序列,我们考虑6个算符(均值、最大值、最小值、标准差、方差和中位数)来整合蛋白质序列的特征,得到每个噬菌体/宿主的162维特征向量。
数据增强模块
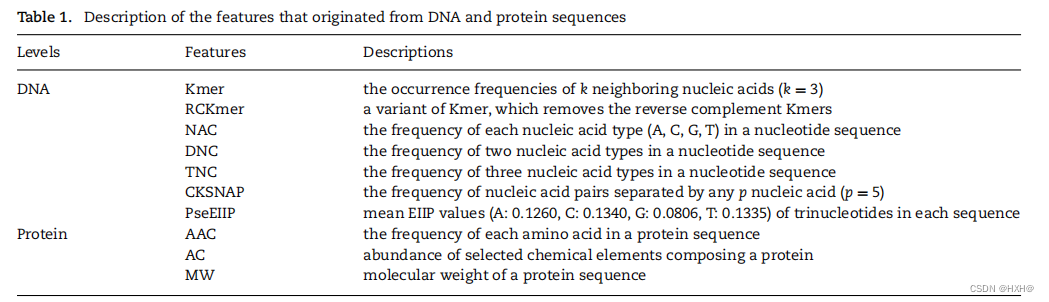
GAN 是一种新型的生成模型,旨在通过精确学习真实样本的潜在分布来生成高质量的伪样本。该模型在的多个领域都得到了广泛的关注,并取得了优异的性能。在本研究中,我们使用GAN来解决模型训练数据集中PHIs数据的稀缺性问题。我们首先将真实的阳性样本设置为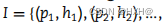
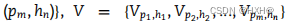
,以代表这些样本的特征向量,由上述噬菌体和宿主的DNA和蛋白质特征组成。M和n分别表示噬菌体和宿主的数量。将这些正样本的特征向量(V)输入GAN,生成高质量的样本伪特征向量,其中GAN由两个神经网络(生成器和鉴别器)组成,相互“对抗”以学习真实样本的分布。一个网络(生成器)试图通过五个完全连接的层来生成伪样本(公式1)。另一个网络(鉴别器),它由四个完全连接的层(公式2)组成,试图区分一个给定的样本是否是真实的。每个网络的任务越来越好,直到达到平衡,生成器不能做出更好的样本,鉴别器不能分离真实样本和伪样本。
其中V为伪样本的特征向量,Oge为生成器输出,Odi为鉴别器输出,FCl(或FCr)表示具有LeakyReLU(或ReLU)激活函数的MLP,FCt表示具有Tanh激活函数的MLP。
我们使用KNN(k = 1)和留一交叉验证(LOOCV)进行分类器双样本测试(C2ST)方法来区分真实样本和伪样本。C2ST方法涉及到接受或拒绝P等于Q的零假设(其中P和Q是两个大小相等的样本集的分布)。如果接受零假设,则预测保留样本的二元标签的分类精度将接近概率水平(即0.50)。因此,当LOOCV下KNN分类精度最接近0.50时,真实样本和伪样本无法区分。最后,我们选择难以区分的伪样本来放大我们的数据集,用表示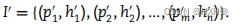
上述处理用于扩增我们的数据集中的阳性样本。由于所有未知的PHIs都是候选阴性样本,并且远远多于阳性样本,所以我们从这个候选集合中随机选择阴性样本,以确保阳性样本和阴性样本的数量相等。最后,将真实阳性样本、选择的阴性样本和伪阳性样本相结合,构建增强数据集,对预测模型进行训练。
PHI预测模块
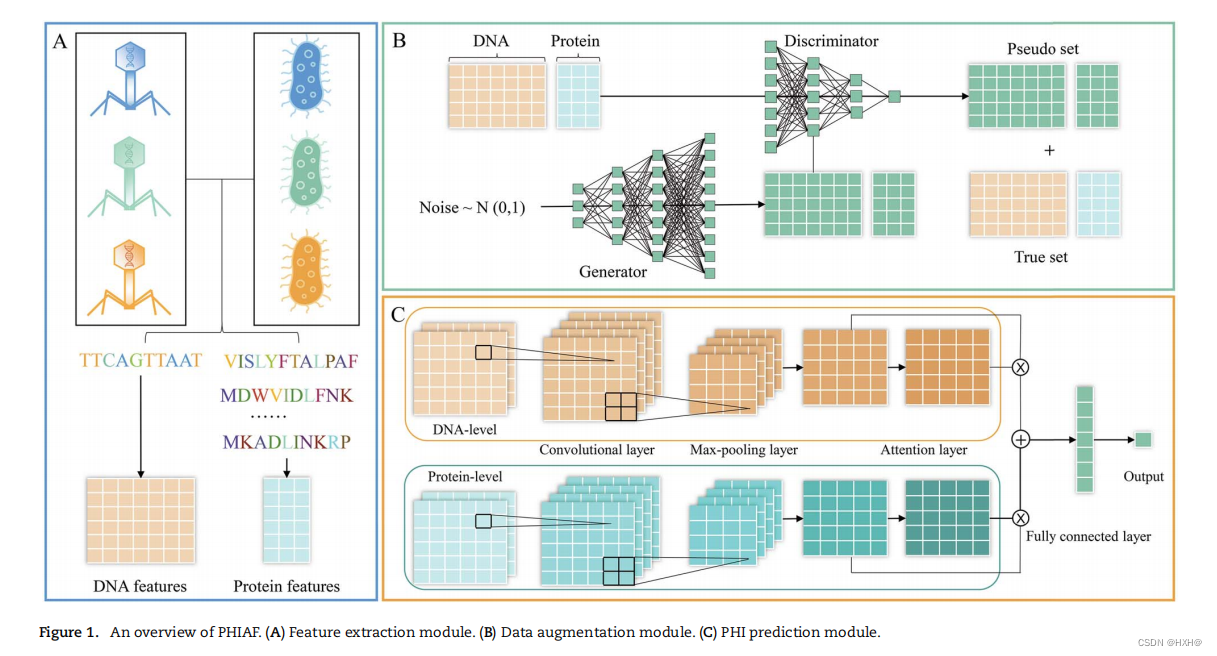
基于增强的数据集,我们利用噬菌体和宿主的DNA和蛋白质序列衍生的特征来建立预测模型。
如前一节所述,我们对每个噬菌体/宿主有两种类型的特征载体,它们分别从DNA和蛋白质序列中提取。由于序列衍生的特征通常具有复杂的短期/长期依赖关系,我们将DNA/蛋白质特征向量重塑为“图像”,以捕捉它们的维度[46]之间的复杂关系。我们首先采用Min-Max归一化,将特征向量的值归一化到0到1的范围。设N表示DNA/蛋白质的特征向量的维数;然后,我们通过按行放置值,将特征向量重塑为n×n‘图像’,其中n满足条件:(n−1)×(n−1)<×和n≤n×n。当N < n×n时,我们通过对剩余的n×n−N条目添加零来实现填充。最后,我们得到了每个噬菌体(或宿主)的DNA和蛋白质序列衍生的特征矩阵,分别记为分别为 MP d和MP p(MH d和MH p)
然后,我们构建了一个双层结构(DNA和蛋白质水平),从DNA和蛋白质特征矩阵中提取更深层次的特征。在dna水平上,我们将噬菌体和宿主的dna衍生特征矩阵跨通道堆叠形成组合矩阵,然后将组合矩阵输入两层CNN,生成具有更有意义信息的特征图Od。CNN包括一个卷积层和一个最大池化层。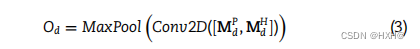 其中,Conv2D和MaxPool分别表示卷积层和最大池化层,[·,·]表示跨通道的堆叠。类似地,噬菌体和宿主的蛋白质衍生特征矩阵跨通道组合,并输入到同一CNN,在蛋白质水平上生成特征图
其中,Conv2D和MaxPool分别表示卷积层和最大池化层,[·,·]表示跨通道的堆叠。类似地,噬菌体和宿主的蛋白质衍生特征矩阵跨通道组合,并输入到同一CNN,在蛋白质水平上生成特征图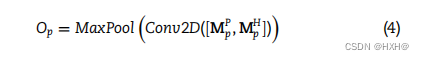
注意机制旨在模仿人类大脑的行为,选择性地集中在一些重要的部分,而在机器学习任务[47]中忽略其他部分。我们拥有的dna和蛋白质水平特征可能对PHI预测做出不同的贡献。因此,我们在模型中引入了一种注意机制,添加了一个注意层来捕获重要的特征,然后整合这些特征。在注意层,将DNA和蛋白质水平的特征图(Od和Op)输入到一个全连接的层中,分别计算权重向量(αd和αp);然后,将特征图乘以相应的权重向量。最后,注意层的输出计算如下: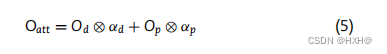
其中⊗是元素级乘法。最后,将注意层的输出输入到两层MLP,以产生样本成为PHI的概率。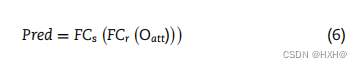
其中FCs是指具有Sigmoid激活功能的MLP
PHIAF优化
在PHIAF的数据增强和PHI预测模块中有几个重要的超参数。在数据增强模块中,我们将发生器不同全连接层中的神经元数量分别设置为128、256、512、1024和1004,将鉴别器中的神经元数量分别设置为512、256、128和1。在PHI预测模块中,我们在卷积层中设置32个滤波器,一个滤波器大小为3×3,将DNA和蛋白质水平的最大池层大小分别设置为3×3和2×2。这两个最大池化层的大小差异确保了Od和Op的维数相等。我们还考虑学习率(可选值为0.1、0.01、0.001和0.001和0.0001)、批大小(可选值为8、16、32、64和128)、辍学层的损失率(可选值包括0.25、0.5和0.75)和完全连接层的神经元数量(可选值包括16、32、64、128、256和512),以确定最佳参数。给定不同参数的模型性能见补充材料第5节。
此外,我们在PHI预测模块中利用辍学层和批处理归一化层来防止过拟合,提高的泛化性。对数据增强模块和PHI预测模块进行了独立训练。此外,我们采用瓦瑟斯坦损失加梯度惩罚作为数据增强模块的损失函数,并对PHI预测模块使用二元交叉熵损失函数。以上所有的损失函数都由Adam优化器进行了优化。
结果
性能评价
在本研究中,我们采用5倍交叉验证(5-CV)来评价PHIAF的性能。我们数据集中的所有样本被随机划分为5个大小相等的子集。交叉验证过程重复5次,每个子集依次用作测试集,其余四个子集用作训练集。最终的5-CV结果是通过平均5个测试集的结果而产生的。我们采用几种常用的评估方法来评估PHIAF的性能和最先进的方法,包括特异性(Spe)、灵敏度(Sen)、准确性(Acc)、f1评分(F1)、受试者工作特征曲线下面积(AUC)和精确召回曲线下面积(AUPR)。这些措施的计算方法如下: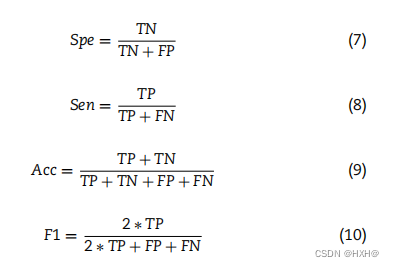
其中,TP(TN)表示在预测中正确分类的阳性(阴性)样本的数量,而FP(FN)表示错误识别的阴性(阳性)样本的数量。
与最先进的方法进行比较
为了证明PHIAF的有效性,我们将其与以下最先进的方法进行了比较,包括基于DNA序列的算法和基于蛋白质序列的方法。
VHM 是一种基于DNA序列的方法,它计算噬菌体与宿主的寡核苷酸频率模式之间的距离,并基于该距离获得PHIs的可能性。
WIsH 是一种基于DNA序列的方法,为每个宿主训练一个Markov模型,并计算所有噬菌体相互作用的概率。
PHP 是一种基于DNA序列的方法,利用病毒和宿主基因组序列之间的k-mer频率来预测PHIs。
RF、SVM、KNN和MLP是被广泛使用的机器学习分类器;Leite等人。利用蛋白质序列衍生的特征和这些分类器来预测PHIs。
PredPHI是一种基于蛋白质序列的方法,在CNN框架下预测PHIs。
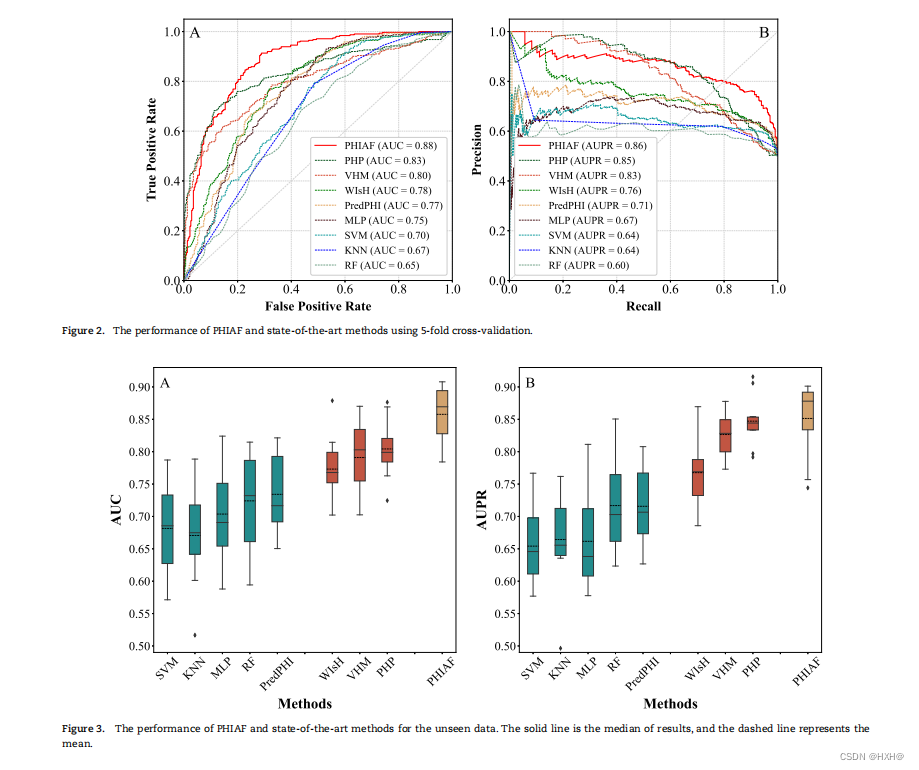
我们首先使用5-CV来比较PHIAF和这些方法。如图2所示,PHIAF在AUC和AUPR方面都优于所有的比较方法。一般来说,基于DNA序列的方法(VHM、WIsH和PHP)比基于蛋白质序列的方法(RF、SVM、KNN、MLP和PredPHI)效果更好,表明来自DNA序列的信息可能在PHI中发挥更重要的作用。我们的PHIAF模型融合了来自DNA和蛋白质序列的信息,优于基于DNA序列或蛋白质序列的模型,在AUC和AUPR方面平均分别提高了13.63%和14.75%。
模型对看不见数据的预测能力也很重要。因此,我们在我们的数据集中随机选择三分之一的噬菌体和宿主,并使用它们和它们之间的相互作用作为一个测试集。剩余的噬菌体和宿主及其相互作用被用于训练预测模型。在这个实验设置下,测试集中的噬菌体或宿主不包括在其中训练集,它们被当作看不见的数据。我们基于训练集对预测模型进行训练,然后将训练后的模型应用于测试集,以评估预测模型对看不见的数据的性能。为了避免评价偏差,我们重复训练和测试10次,并采用平均/中位数的表现。如图3所示,PHIAF优于其他方法,产生更高的平均AUC(0.86)和AUPR分数(0.85),以及合理的标准差,AUC和AUPR评分的中位数分别为0.88和0.87。与5-CV的结果相似,基于DNA序列的方法比基于蛋白质序列的方法产生更好的结果。此外,比较5-简历的结果和测试看不见的数据表明,我们的方法实现非常相似的性能在这两种情况下(AUC和AUPR相差2%和1%),这些结果表明,我们提出的方法是健壮的,可以执行看不见的数据,表明PHIAF是一个有前途的工具来识别艾滋病从序列数据。
消融研究
以上比较说明了PHIAF的有效性,PHIAF的成功是其设计的结果:一个基于GAN的数据增强模块生成高质量的伪样本,一个融合DNA和蛋白质序列衍生特征与注意机制的PHI预测模块。在这里,我们进行了一项消融研究来详细阐述这些成分的贡献。我们考虑以下PHIAF的变体:
- PHIAF-D是一种不使用DNA水平特征的变体。
- PHIAF-P是一种不使用蛋白质水平特征的变体。
- PHIAF-A是一种不使用注意层的变体。
- PHIAF-G是一种不使用伪样本的变体。
表2显示了5-CV下PHIAF及其四种变异的结果。当删除任何组件时,PHIAF的性能都会下降,这意味着所有组件都是关键的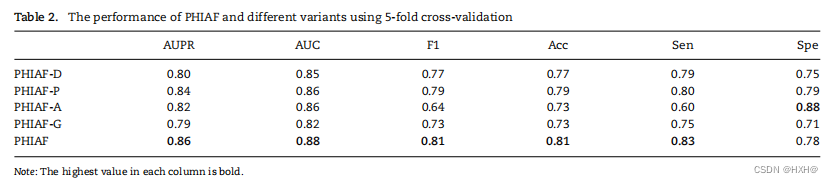
为PHIAF的。PHIAF-G的性能下降幅度最大,AUC和AUPR分别下降6%和7%,其次是PHIAF-D(AUC和AUPR分别下降3%和6%),PHIAF-A(AUC和AUPR分别下降2%和4%)。PHIAF和PHIAF-G的结果比较表明,伪样本的使用有效地提高了PHI的预测。此外,PHIAF-D和PHIAF-P的比较表明,DNA序列衍生的特征比蛋白质序列衍生的特征更能有效地预测PHI。去除注意层也会导致较差的表现,这表明必须考虑到特征上的差异。
讨论
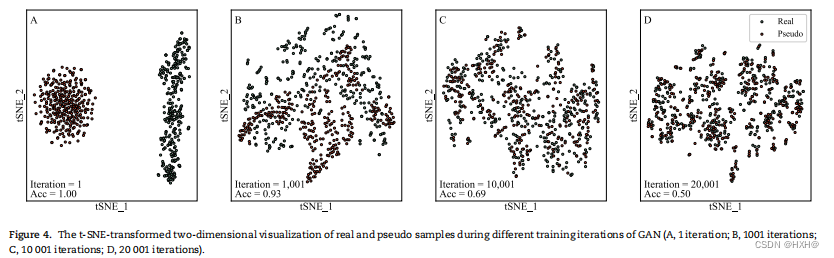
消融研究表明,PHIAF的主要成分对PHI的预测有重要贡献。进一步地,我们从三个方面分析了PHIAF。为了证明真实样本和伪样本是难以区分的,并且伪样本可以作为训练正样本,我们使用t分布随机邻域嵌入(t-SNE)来可视化真实样本和伪正样本在不同训练迭代下的二维分布(图4)。在第一次迭代中(图4A),真实样本分布在离伪样本较远的地方,导致LOOCV精度为1.00,说明生成器没有学习到真实样本的分布。如图4B所示,生成器开始捕获真实样本的特征,经过1000次迭代后,鉴别器不能完全区分真实样本和伪样本(LOOCV精度为0.93)。然后,分布继续逐渐收敛(图4C,LOOCV精度达到0.69)。经过20 000次迭代后,生成器和鉴别器达到平衡,真实样本和伪样本遵循相似的分布(图4D,LOOCV精度为0.50)。因此,我们生成了高质量的伪阳性样本,用于数据扩充。
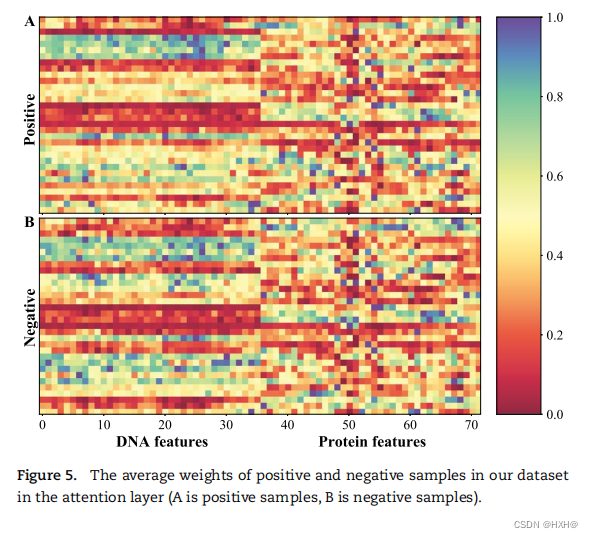
进一步,我们分析了赋予不同特征的注意层的权重,以研究由注意机制学习到的特征的重要性。从图5A和B可以看出,正样本和负样本的注意权重分布相似,说明一些特征在正样本和负样本中起着相同的重要作用。此外,我们还比较了在DNA水平和蛋白质水平上的注意权重,其中蛋白质水平的特征通常被分配的权重低于DNA水平的特征。这些结果证实了DNA水平的特征在PHI预测中比蛋白质水平的特征更重要,CNN中的注意层不仅提高了预测性能,而且可以根据不同特征的重要性有效地分配权重。
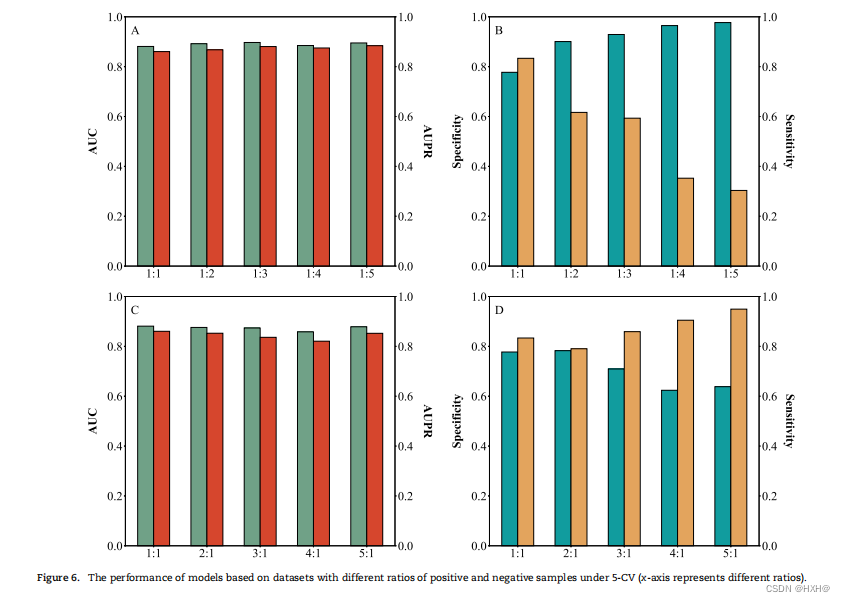
最后,我们基于不同正负样本比例(1:1、1:2、1:3、1:4、1:5、2:1、3:1、1、4:1和5:1)的数据集建立PHIAF模型,分析负或伪正样本的数量如何影响PHIAF的性能。如图6A和6C所示,PHIAF在不同数据不平衡比率的数据集上产生相似的AUC和AUPR评分(在2%内变化)。随着阴性样本数量的增加(图6B),PHIAF获得了较低的敏感性(从0.83下降到0.30)和较高的特异性(从0.78增加到0.98)。相比之下,当伪阳性样本数量增加时,PHIAF产生更高的敏感性(从0.79增加到0.95)和更低的特异性(从0.78下降到0.62)(图6D)。这些结果表明,不平衡的数据集并不能显著提高PHI预测的性能,而样本很可能被预测为负/正。
研究实例
在本节中,我们将进行一个案例研究来估计PHIAF预测未知新PHI的能力。我们首先使用2021年1月1日之前出现在NCBI中的所有噬菌体和宿主的已知相互作用来训练PHIAF,以识别剩余的噬菌体和宿主之间的所有成对(其余的代表2021年1月1日之后在NCBI中出现的噬菌体和宿主)。然后,我们根据预测评分对PHI进行排序,并搜索最新发表的文献,以验证预测的PHI是否已被生物学实验证实。我们在表3中列出了这些预测的PHI(按预测分数排序);其中四对最近出版的文献得到了验证。例如,[的分析结果描述了Kokobel1(NCBI登录号: NC_052979)可以杀死普罗维登西亚(NCBI登录号: NZ_CP029736)和普罗维登西亚(NCBI登录号: NC_017731)的一些菌株。Zhan等[55]报道了5种噬菌体感染NCBI DSS-3(NCBI登录号: NC_003911),其中一种为vB_RpoS-V16(NCBI登录号: NC_052969)。本案例研究的结果表明,PHIAF可以帮助识别新的PHIs,并缩小进一步生物学实验的候选范围。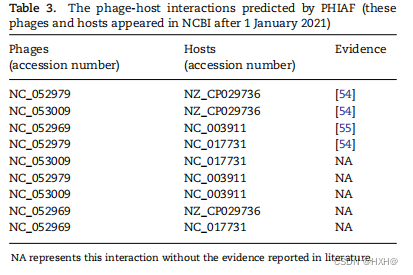
结论
抗生素的过度使用给细菌性疾病的治疗带来了一些严重的挑战。噬菌体治疗作为治疗细菌性疾病最有前途的替代抗生素之一,受到了广泛的关注。确定PHI对于理解噬菌体是否可以用于治疗细菌性疾病极为重要。在本研究中,我们提出了一种PHIAF的PHI预测方法,该方法利用GAN生成高质量的伪样本,融合来自DNA和蛋白质序列的特征以获得更好的性能,并使用注意机制来提供预测模型的可解释性。通过5-CV与最先进的方法进行比较表明,PHIAF实现了最好的PHI预测(在AUC和AUPR方面,性能平均提高约为13.64%和14.75%)。此外,消融研究说明了PHIAF各组成部分的贡献,数据增强模块对预测模型的贡献最大。并通过案例研究证明了该方法的可行性。实验结果表明,PHIAF是一种很有前途的识别PHI的工具。
尽管我们的模型具有良好的预测性能,但仍有一些限制有待解决。例如,我们所设置的多个超参数的初始值和范围都来源于以前的研究,仅在有限的实验中粗略确定。在未来,我们可以通过训练更多的预测模型来获得最优的超参数。此外,PHIAF作为一种旨在预测PHIs的网络体系结构,可以用于处理生物信息学中其他基于序列的分类任务。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









