







由于PROTACs的构效关系比较模糊,当前暂时没有PROTAC理想的理性设计与药效评价计算方法。上海科技大学一研究团队引入了DeepPROTACs,以帮助设计有效的PROTACs分子。它可以根据给定的靶蛋白和E3连接酶的结构来预测PROTAC分子的降解能力。

背景介绍
传统疗法依靠小分子抑制剂作为作用模式(MOA)实现占位驱动药理学。该方法存在无法处理不可成药的靶点、脱靶毒性、不良副作用、耐药性等缺点。单克隆抗体和RNA干扰(RNAi)方法开始补充小分子抑制剂方法,但仍存在抗体难以穿过细胞膜,RNAi分子本身的口服生物利用度和组织分布较差等弱点。鉴于以上情况,本文采用PROTACs技术作为MOA。
蛋白水解靶向嵌合体(PROTACs)自2001年诞生以来,已成为利用事件驱动MOA的一项十分具有吸引力的技术。PROTAC是一种特异性双功能分子,由一个靶蛋白(POI)配体、一个连接子和一个E3泛素连接酶配体组成。它通过将泛素化机制带到POI附近来促进三元复合物(POI-PROTAC-E3)的形成,驱动泛素从E2泛素结合酶转移到靶蛋白上并与表面的赖氨酸共价结合。随后,泛素化标记的POI被26 S蛋白酶体识别并降解为短肽甚至氨基酸(图1)。
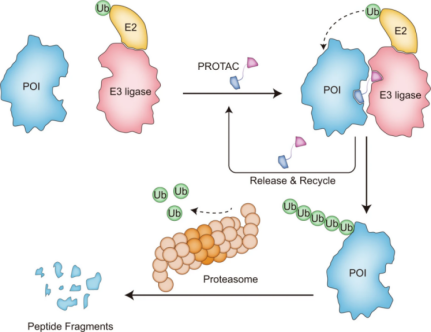
PROTACs的优越性
作为一种新颖且具有发展前景的技术,与现有的处理方法相比,PROTACs具有多种优越的性能。
(1) PROTACs能够调节缺乏经典疏水药物结合口袋或与内源性分子强结合的不可成药靶点。此外,它还可以处理一类与蛋白质相互作用的蛋白质。
(2) PROTACs具有催化作用,因为一旦泛素化过程完成,它们就会从三元复合物中释放出来。由于这种催化性质,PROTACs可以在低暴露下发挥作用,减少脱靶和其他不良影响的可能性。
(3) 使用PROTACs可以避免靶点积累,调节非酶/支架功能,并解决结合口袋周围突变引起的耐药性问题。
(4) 通过应用PROTACs可以提高密切相关的蛋白质的选择性。同源蛋白的活性位点高度保守,而催化核心外的序列和构象可能发生较大变化。泛素转移步骤取决于表面的赖氨酸和泛素的相对位置,因此PROTACs可以利用这种差异来降解特定的靶点。
PROTACs的数据库源
最近,Hou等人发布了一个在线的PROTACs数据库(PROTAC-DB),其中包括2258个PROTACs, 275个弹头(靶向POI的小分子),68个E3配体(招募E3连接酶的小分子)和1099个连接子。此外,本文团队还从其他公共来源收集了更多数据(375个PROTACs,针对30个POIs)。PROTAC-DB为各种各样的PROTAC提供了结合亲和力、降解效率和细胞活性。因此,本文团队可以方便地从这个数据库中获取数据。
DeepPROTACs模型
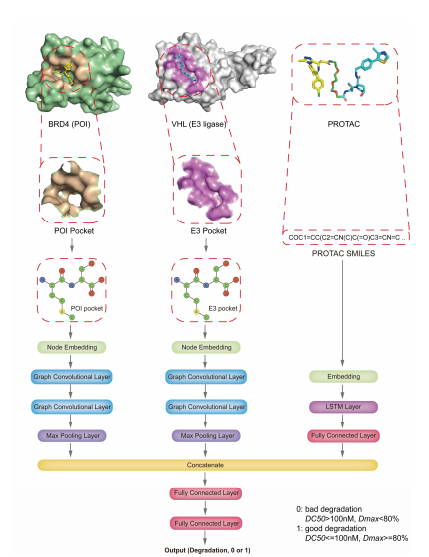
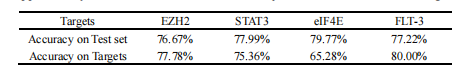
在本研究中,本文引入了一种深度神经网络模型DeepPROTACs,该模型可以基于POI和E3连接酶的结构,有效预测给定PROTACs的降解效果。该框架用给定POI-PROTAC-E3连接酶复合物的不同部分嵌入到单独的神经网络模块中。组件嵌入连接在一起,然后馈送到一个具有两个完全连接层的MLP以获得最终的输出。模型在测试集上的平均预测准确率达到78%左右,ROC曲线下面积(AUROC)达到0.85左右。本文通过使用一批VHL破坏雌激素受体(ER)的PROTACs进一步验证了该模型。在这16个PROTACs中,该模型成功预测了11个PROTAC的降解能力,预测精度达到68.75%。对于最近报道的其他PROTAC靶点(EZH2、STAT3、eIF4E和FLT-3),准确率在65%~80%之间。所有这些结果都证明了该DeepPROTACs模型具有良好的泛化能力。
DeepPROTACS的网络架构
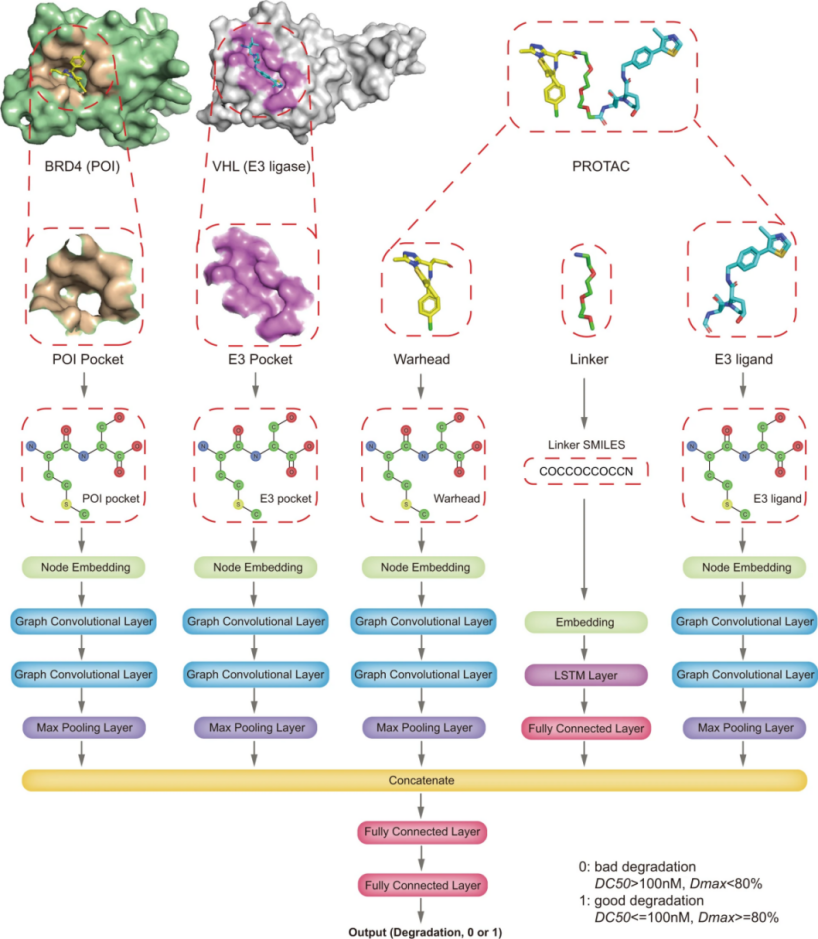
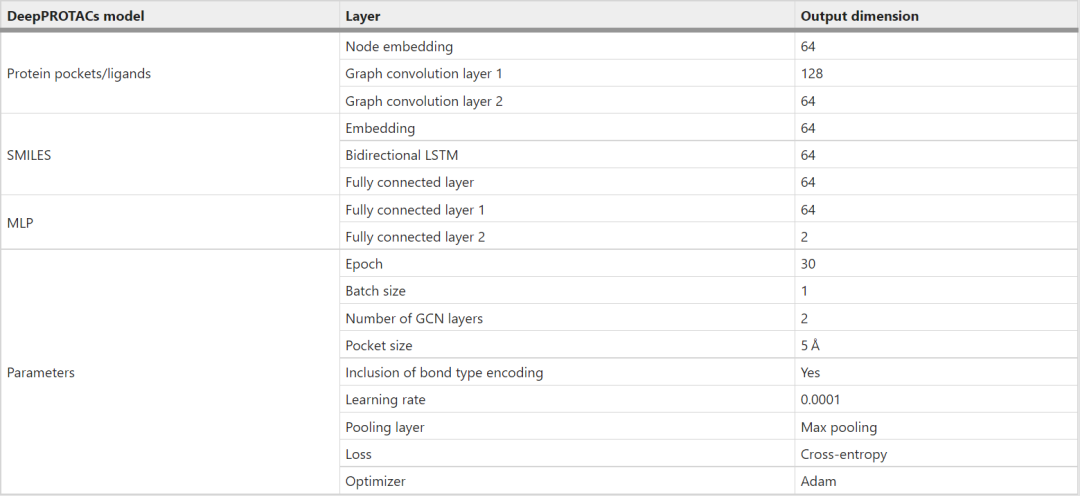
本文用PyTorch和PyTorch几何框架实现了整个网络。在本文的实验中,所有的GCNs都由两个图卷积层和一个最大池化层组成。每个最大池的输出口袋/配体表示的维数为64。此外,将连接子SMILES的编码输入嵌入层以获得分布式表示。然后,该嵌入层的输出依次在64 个节点和全连接层的双向 LSTM 层中馈送。连接子SMILES网络模块的输出表示也具有64维。该连接子网络的结果与口袋/配体网络的结果相连接,然后被送入具有两个完全连接层的MLP以获得最终输出。该网络采用渗漏整流线性单元(Leaky ReLU)作为激活函数。注意,POI口袋和E3口袋的GCNs的重量是共享的,弹头和E3配体的GCNs的重量也是共享的。表1列出了 DeepPROTACs 模型中使用的每层的详细输出尺寸和超参数。
表1 DeepPROTACS模型的尺寸和参数
为了将该模型与基线方法进行比较,本文利用自动交叉协方差(ACC)作为蛋白质的特征,进一步训练了支持向量机(SVM)和随机森林(RF)等几种传统ML模型。配体由分子访问系统(MACCS)键或摩根指纹表示。ACC将蛋白质序列转换为18位向量,而MACCS密钥和Morgan指纹分别代表一个166位向量和1024位向量的小分子。三元配合物的表征是通过连接靶蛋白、E3连接酶和PROTAC分子的特征来构建的。在Scikit-learn包中建立支持向量机模型,设核为线性,正则化参数C为1;在Scikit-learn包中还构建了射频模型,并将估计量的数量设置为100个,最大深度为5。
模型参数的优化
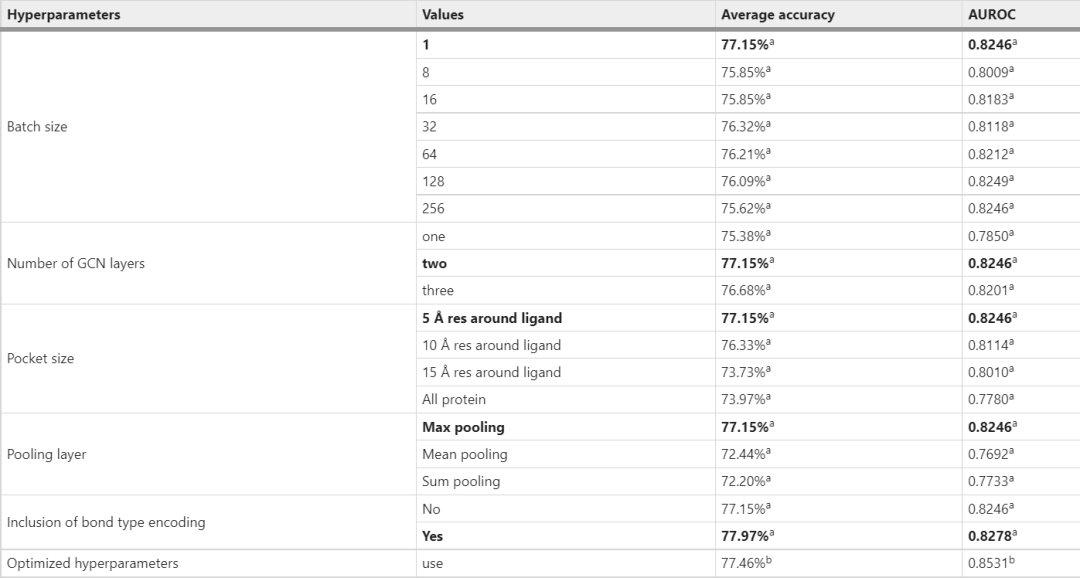
每个实验中,整个数据集以8:1:1的比例随机分为训练集、验证集和测试集。最终,该模型在验证集上达到了 77.15% 的平均准确率。本文还研究了GCN层数的影响,并观察到层数为2的最佳性能(表2)。通过逐渐扩大口袋大小来研究口袋大小对模型的影响。如表2中所列,使用5 Å口袋尺寸的型号性能最佳。5 Å大小的口袋仅包含配体周围的第一和第二配位壳残基,这些残基在POI/弹头和E3/配体结构中已知。此外,加入最大池化层和键类型编码可提高模型的性能(表2)。最终模型由表1中列出的优化参数和 Adam 优化器训练学习率为 0.0001,β1 为 0.9,β2 为 0.999。
表2:验证集上DeepPROTACs模型超参数的优化
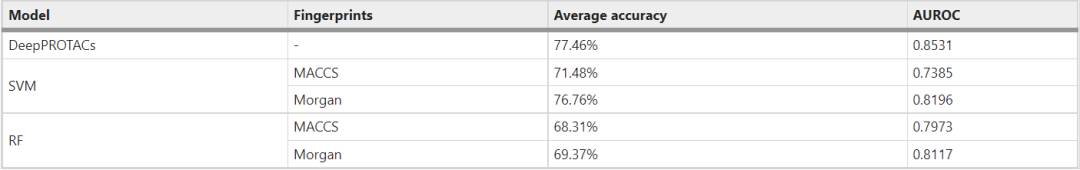
经过三次重复训练,DeepPROTACs模型的平均准确率为77.46%,AUROC为0.8531(表3)。与使用不同指纹的SVM和RF模型的指标(表3和图3)相比,这是一个令人印象深刻的成就。与MACCS密钥相比,带有Morgan指纹的SVM模型的性能有了很大的提高。但与GCN模型相比仍有一定差距,特别是在AUROC方面。RF模型的平均准确率接近70%,AUROC在0.80左右,均不如SVM和GCN模型。在接下来的实验中,本文采用8:2(训练集:测试集)的划分,在更大的测试集上充分检验DeepPROTACs模型的预测能力。结果表明,DeepPROTACs在测试集上保持了较高的性能(准确率为77.95%,AUROC为0.8470)。
表3:DeepPROTAC、SVM和RF模型在测试集上的评估结果
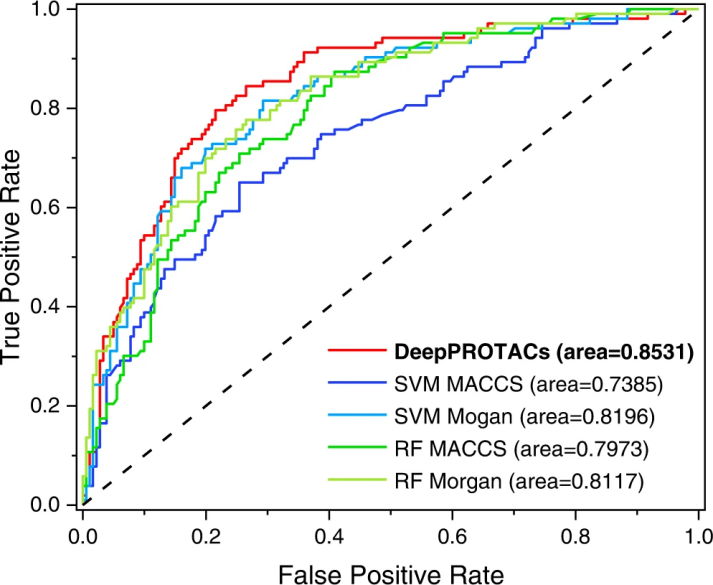
为了进行比较,本文构建了两个替代模型,分别将整个PROTAC分子视为图和SMILES(图4和图5)。然而,这两个模型对测试集的预测精度分别为68.08%和76.25%,低于DeepPROTACs模型。在DeepPROTACs中,不仅可以降低图表示中邻接矩阵的稀疏性,还可以揭示连接子与降解效果之间的隐藏映射。因此,丢弃这两个替代模型,接下来的实验侧重在DeepPROTACs模型上。

架构验证和数据平衡
本文在DeepPROTACS模型上进行了消融实验,以验证当前的网络架构。如图6a所示,从当前架构中消除连接酶口袋,E3配体,POI口袋或弹头(消融项目:2,3,4,5)确实会削弱GCN模型的性能。删除linker输入(已消融项:6)也会导致性能下降。此外,与单个项目的消融相比,去除双项目,例如连接酶口袋/E3配体或POI口袋/弹头(消融项目:7,8)进一步降低了预测精度和AUROC。简而言之,这些实验证明了当前DeepPROTACS模型中每个部分的不可或缺性。此外,通过使用三种不同的训练/测试拆分重复了DeepPROTACS模型的训练过程。对于每次拆分,整个数据集以 8:2 的比例随机分为训练集和测试集。在每次拆分的情况下,模型都经过三次训练。每个拆分的评估指标非常相似,说明了本文的GCN模型的稳健性和可重复性。
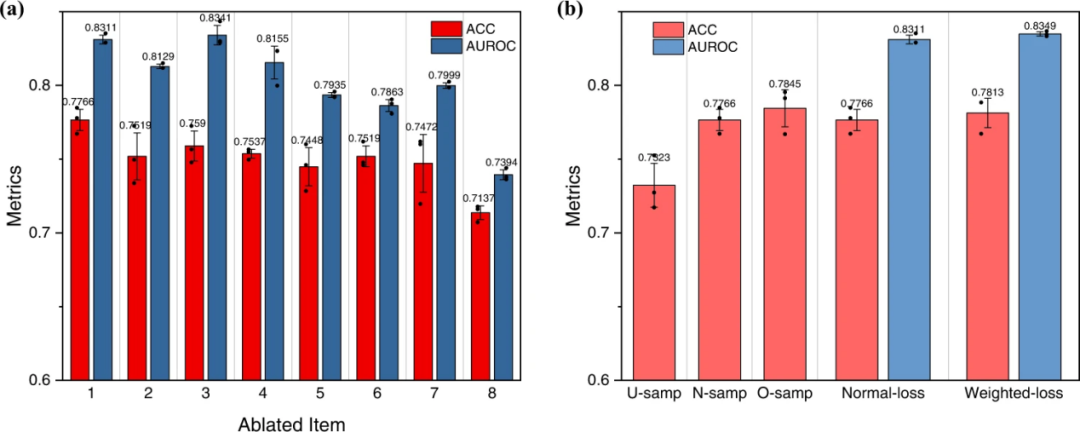
图6:在3个独立实验中对N = 2832个生物独立样品进行DeepPROTACs模型验证和数据平衡检查
本文发现,PROTAC分子的活性分布在不同对靶蛋白和E3连接酶之间差异很大。因此,在单个特定的靶蛋白和E3连接酶之间几乎不可能实现数据平衡。相反,本文试图研究数据平衡对整个数据集的影响。在每种采样方法中,本文对模型进行了三次训练,得到了平均预测精度和AUROC。本文的实验结果表明,过采样方法的性能最好,其次是正态采样和欠采样(图6b)。因为过采样充分利用了有限的数据,而欠采样浪费了一些数据资源。此外,加权损失也被用来作为另一种方法来改善数据不平衡的影响。这些结果说明了数据不平衡的问题在本研究中确实存在,而试图解决这一问题的努力只能在一定程度上提高模型的性能。因此,考虑到性能和计算成本之间的平衡,本文采用默认设置(正态采样和正态损失)来训练最终的DeepPROTACs模型。
DeepPROACs模型以半降解浓度(DC50)和最大降解水平(Dmax)为依据,将降解率的预测简化为二分类问题。
DeepPROTACs的评估
为了验证DeepPROTACs的预测能力,本文构建了一个包含16个使用VHL E3连接酶降解ER的PROTACs的实验数据集(图7)。
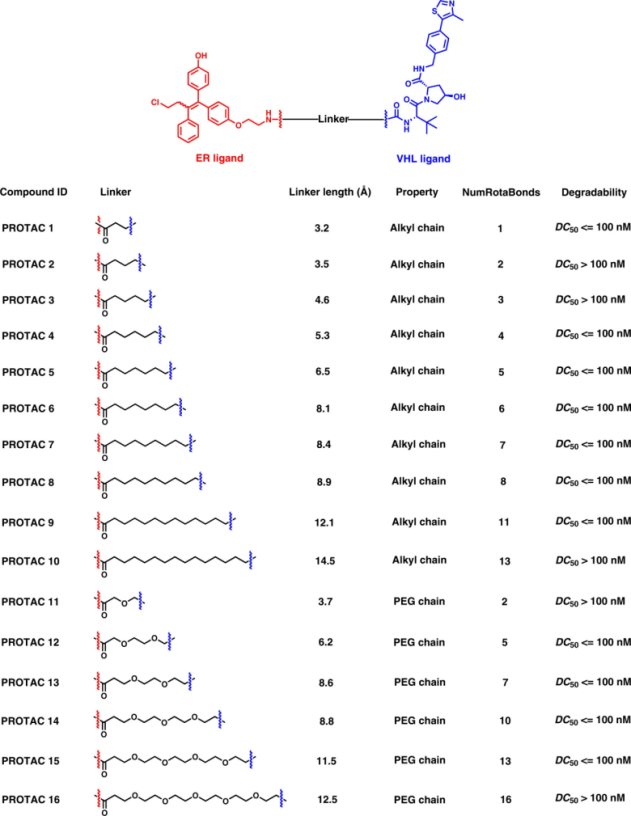
蛋白质印迹数据(图8和表4)显示,11种化合物(PROTAC 1、PROTAC 4 ~ 9、PROTAC 12 ~ 15)在16小时内诱导浓度低于100nM的ER降解非常有潜力。因此,它们被认为是良好的降解剂。其他5种PROTAC (PROTAC 2 - 3, PROTAC 10 -11, PROTAC 16)在指定浓度下降解ER的效果较差或无效,这意味着它们属于不良降解剂。在T-47D细胞系中对PROTAC 8及其阴性类似物PROTAC 8N(含VHL配体的非活性异构体)进行VHL/ER结合试验和蛋白质印迹分析。

表4 蛋白质印迹数据的密度定量顺序
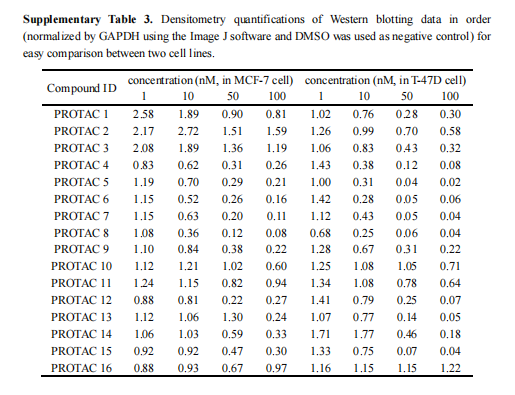
根据图9a的热力学数据计算发现PROTAC 8N与VHL之间几乎没有结合。此外,在指定的浓度(100/1000 nM)下,PROTAC 8几乎完全降解ER,而PROTAC 8N不能降解ER(图9b)。这些结果证实了ER PROTACs与VHL连接酶和ER蛋白的结合,表明ER的降解确实是通过泛素-蛋白酶体途径实现的。DeepPROTACs模型成功预测了16种PROTACs中11种化合物的降解标签(表5),预测准确率达68.75%。
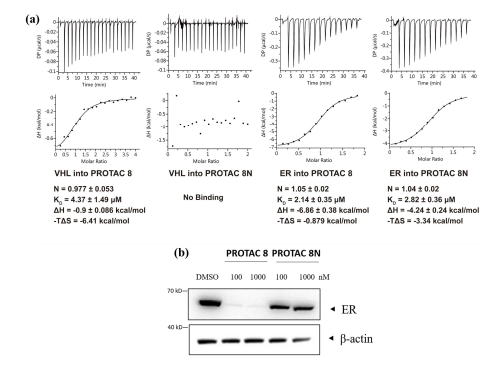
表5 模型预测结果
将表6的靶标(EZH2、STAT3、eIF4E和FLT-3)视为新靶标,以进一步检验模型的泛化能力。测试集上的所有预测精度都非常相似,在 77% 的值左右波动。特定目标的准确度在65%到80%之间变化,说明本文的模型具有良好的泛化能力。
表6 模型预测准确性
计算建模:ER PROTACs
化合物PROTAC 2,PROTAC 6和PROTAC 10分别被证实是坏降解剂,好降解剂和坏降解剂。它们的接头长度计算为3.5、8.1和14.5 Å,分别代表短、中、长烷基接头(图7)。因此,他们被选中通过PRosettaC和分子动力学(MD)模拟构建三元复合物。
如图10所示,根据ER蛋白的位置对这些复合物最具代表性的结构进行了排列。本文根据模型发现不同的连接子导致了三元复合物的不同构象,从而改变了整个CRL结构,改变了表面赖氨酸残基对Ub的可及性。尽管CRL的泛素化区在一定程度上是大而灵活的,但这仍可能对降解能力产生深远的影响。中等长度的linker具有良好的降解性,因为它们为三元复合物提供了灵活性和稳定性。
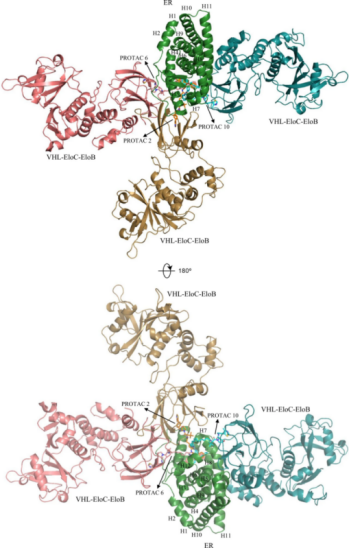
图10:从两个视图构成的由ER,VHL-EloC-EloB和PROTAC(PROTAC 2,6和10)构成的三元复合物的计算模型
总体而言,将测试集上 77.95% 的平均准确率和 0.8470 AUROC 与 ER 实验数据集和其他新靶点的65%至80%准确率相结合,DeepPROTAC 模型能够预测提供 POI、E3 连接酶和 PROTAC 结构的新型 PROTAC 的降解能力。
总结
本文提出了一个DL模型—DeepPROTACs,以解决设计有效的PROTAC分子(尤其是连接子)的困难。DeepPROTACs不仅为PROTACs的设计提供了一种高通量筛选方法,而且为AI与药物发现的融合提供了一种研究范式。
参考资料
Li, F., Hu, Q., Zhang, X. et al. DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs. Nat Commun 13, 7133 (2022).
https://doi.org/10.1038/s41467-022-34807-3


















 1184
1184

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











