 大家好,我是喜欢看CRISPR花式变戏法的无花果。
大家好,我是喜欢看CRISPR花式变戏法的无花果。前段时间为大家介绍了dCas9技术和CRISPR/Cas9文库筛选,发现大家都比较感兴趣,于是我决定一不做二不休,干脆整一个CRISPR系列,具体讲一讲CRISPR衍生出的各种玩法。今天要介绍的是最近发表在
Nature Biotechnology上的一项技术,
利用CRISPR系统定向地将m6A加到某个mRNA的特定碱基上,实现m6A的定点编辑。有了这项技术,以后就可以研究
单个碱基位点
m6A的功能啦!是不是很神奇?赶紧来看一看吧~

Q&A
Q:为什么要发明这项技术?
A:以往我们研究m6A时,往往是进行
整体水平的m6A检测,或者通过MeRIP检测某个RNA片段上是否有m6A位点,之后再通过敲减/过表达m6A相关蛋白研究m6A。然而,如果想要研究
单个位点的m6A功能,就不好实现。
Q:这项技术的原理?
A:m6A甲基化的调控分为三个部分:甲基的写入(
Writer)、擦除(
Eraser)和读取(
Reader)。相信熟悉m6A的小伙伴都知道,这三个过程分别对应三种蛋白,比较常见的Writer蛋白有
METTL3/METTL14复合体,Eraser蛋白有
ALKBH5、FTO,Reader蛋白有
YTHDF1-3等。本文就是利用m6A writer METTL3/METTL14复合体的
m6A写入功能,以及CRISPR系统的
引导编辑功能,实现对RNA单个碱基位点的m6A编辑。
Q:文中使用的CRISPR系统和传统的CRISPR/Cas9有什么区别?
A:文中所使用的dCas13蛋白即
没有切割活性的Cas13(和之前介绍过的dCas9很类似,具体请戳【前沿技术】CRISPR/dCas9技术带你玩转分子互作),与dCas9不同的是,dCas13靶向的是单链的RNA,而非双链的DNA。
Q:这项技术的优点?
A:如前所述,使用CRSPR/dCas13融合METTL3/14表达的系统能够定点对RNA进行m6A编辑,实现了m6A的单位点研究,并且文中提到该系统脱靶率低,特异性高,这在其他技术手段上是难以实现的。
Q:这项技术容易做吗?
A:这项技术所需要的实验条件要求不高,难点其实是在
sgRNA和dCas13-METTL3/14系统
的设计,操作是常规的载体构建和转染细胞,以及m6A位点的检测和验证,从技术上来说是容易上手的,无论是经验丰富的实验能者还是科研小白都可以尝试。
Q:会不会出现CRISPR系统脱靶问题?
A:脱靶是CRISPR面临的最大问题。本文中作者对该系统的脱靶率进行了检测,证实该系统脱靶率低。当然,在不同细胞中的敲除效率和脱靶率可能都会不同,那就可以像作者一样进行
脱靶率的检测,确保系统的
有效性和
特异性。
接下来,让我们来看看作者具体是如何实现的。
Step1:设计并构建CRISPR/Cas13载体。虽然说构建载体是日常实验中几乎每天都要做的,但是这一最简单的实验却可能是决定后续实验成功与否的关键。本文中的Cas13需要融合表达m6A甲基转移酶,还要去除切割活性(
dCas13),保证高效性和特异性,更需要谨慎设计。作者对这一系统的构建经过了以下过程:
①选择合适的甲基转移酶。为了保证系统的特异性,首先要保证Cas融合蛋白结合RNA位点的特异性。因此需要无结合RNA活性却具有催化活性(能转移甲基)的甲基转移酶。作者选择了METTL3,并对其结合及催化活性进行了检测。
②设计RNA靶向融合蛋白系统。首先将METTL3的锌指RNA结合结构域去除(METTL3ΔZF)进一步削弱其RNA结合能力;又将这一改造的蛋白与METTL14与METTL3互作的结构域融合表达作为候选;最后通过连接序列将dCas13和这两种改造的蛋白融合表达(
dCas13-M3,dCas13-M3M14)。
Step 2:使用大肠杆菌进行验证。首先设计dCas13载体中的IPTG诱导型启动子和组成型表达的gRNA,构建包含特定序列的(GGACU)的靶基因转录载体(人工合成以方便检测)。然后将两种构建好的质粒转化大肠杆菌,使用
MeRIP-RT-qPCR和
MeRIP-seq检测IPTG诱导前后的mRNA甲基化情况。dCas13-M3和dCas13-M3M14均能有效对RNA进行m6A编辑。测序结果显示两者的脱靶效率均在可控范围内,且与其甲基化活性相关。
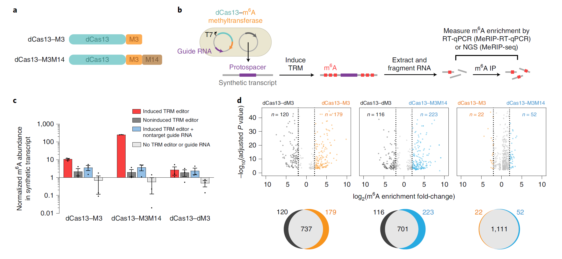
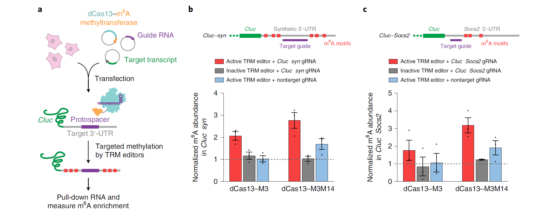
Step 3:在人源细胞中验证系统对外源转录本的m6A编辑作用。作者设计了一段人工合成的目标转录本syn,将其连接在
报告基因Cluc的3‘-UTR端,并且设计了相对应的gRNA,共转染HEK293T细胞。Me-RIP-RT-qPCR检测Cluc syn的m6A水平以说明系统的靶向编辑效率。进一步将合成基因换成Socs2基因的转录本,获得了相同的结果,表明Cas13-M3、Cas13-M3M14能够有效对外源转入的RNA进行靶向位点的m6A编辑。
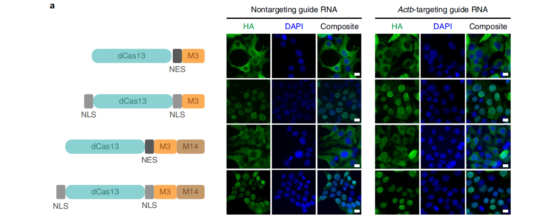
Step 4:设计构建定位于胞质/核的编辑系统。在dCas13-M3/dCas13-M3M14中插入胞质或核表达的
定位序列以及
标签
蛋白,用免疫荧光的方法验证定位(见下图)。
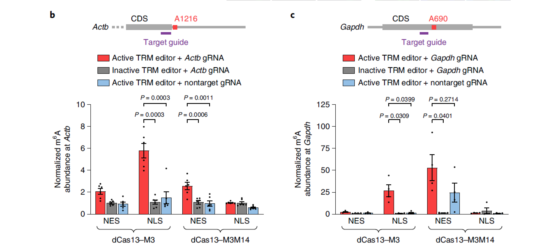
Step 5:在人源细胞中验证系统对内源转录本的m6A编辑作用。设计靶向
Actb A1216和
Gapdh A690位点的gRNA,与dCas13-M3nes/nls、dCas13-M3M14nes/nls共转染HEK293T细胞,Me-RIP-RT-qPCR结果显示这两个转录本的位点可以被加上m6A甲基。
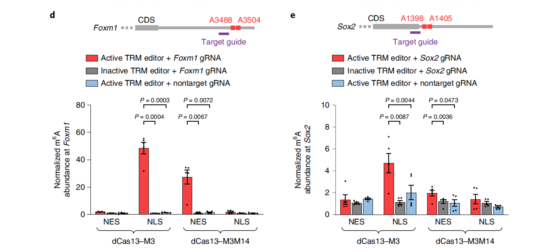
为了进一步检验该系统是否适用于具有生物学意义的基因编辑,作者又选择了两个有代表性的基因
Foxm1(胶质瘤中高甲基化)和
Sox2(甲基化调控干细胞分化)作为靶向转录本。获得的结果与Actb和Gapdh的结果类似。
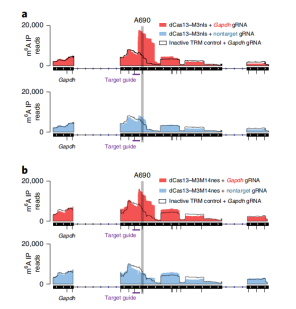
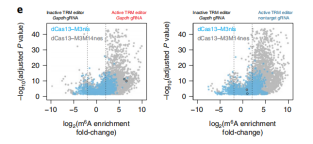
Step 6:靶向RNA甲基化系统的特异性检测。为了检测dCas13-M3/M3M14系统甲基化编辑的
特异性,将gRNA设计为靶向A690上游8个碱基。m6A IP显示被编辑的位点在A690处富集,说明该系统在内源转录编辑上有较好的特异性。
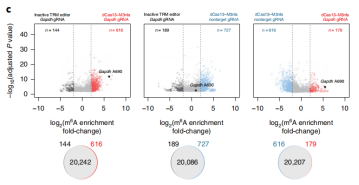
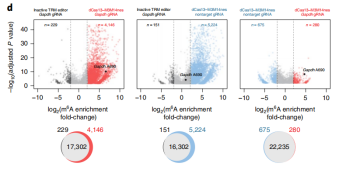
Step 7:脱靶率检测。用Me-RIP-seq检测内源转录本m6A编辑的脱靶率,发现dCas13-M3M14与对照相比差异基因数量要远高于dCas13-M3,表明dCas13-M3脱靶率更低。
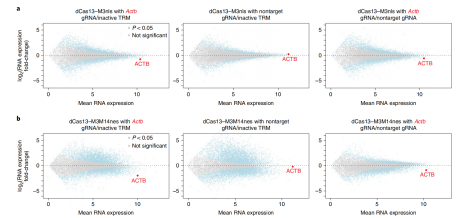
Step 8:验证dCas13编辑系统产生的m6A对靶基因RNA的影响。m6A的修饰往往会影响靶基因的表达量,从而产生生物学意义。为了验证该系统产生的m6A修饰对
靶基因表达量的影响,对靶向Actb A2316位点编辑的细胞进行
RNA-seq。以往研究发现该位点的m6A促进Actb mRNA的降解。转入dCas13-M3/dCas13-M3M14的细胞Actb表达量显著降低。
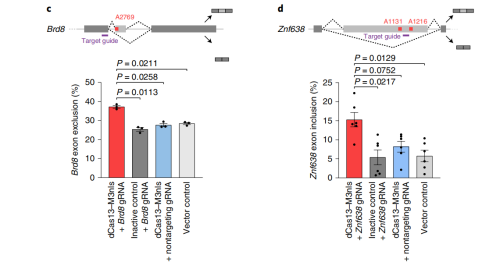
作者又将该系统靶向具有m6A相关
可变剪接位点的
Brd8和
Znf638,结果两个基因RNA的可变剪接均显著升高。
总结其实之前已有报道使用CRISPR/Cas9系统对RNA进行m6A编辑(相关文章发表于nature chemical biology, 全文链接:https://www.nature.com/articles/s41589-019-0327-1),相比较而言,本文使用的dCas13系统操作
更简单,
脱靶率更低,且
使用范围更广(可根据需要选择定位于
核还是
胞质)。相信在不久的将来,除了实现RNA的m6A定点植入外,还能实现m6A的定点擦除,甚至可以实现m6A的定点读取。届时,我们就能真正对m6A展开单个位点的研究。
正在做m6A课题研究的你,不妨试试这一新技术吧。
注:此推文未经许可禁止转载!
阅读推荐:
【前沿技术】CRISPR/dCas9技术带你玩转分子互作
【前沿技术】高水平研究经常用到的基因筛选方法,你Get了吗?
ELISA技术:从技术细节到数据分析演示
用好这个技术,顶刊离我们又近了一步!
【课题套路】一文解读m6A去甲基化酶研究的课题设计思路
如果您或者科室有科研上的困扰
扫码备注:科研思路探讨
























 3万+
3万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









