







 本文介绍了Colabfold,一个简化版的AlphaFold,用于sequence到pdb的预测,适合资源有限的环境。内容包括Colabfold与AlphaFold的区别、安装过程、使用方法及如何处理复合物。此外,还提到了mmseq2的安装和可能的用途。
本文介绍了Colabfold,一个简化版的AlphaFold,用于sequence到pdb的预测,适合资源有限的环境。内容包括Colabfold与AlphaFold的区别、安装过程、使用方法及如何处理复合物。此外,还提到了mmseq2的安装和可能的用途。
啥是colabfold?
sequence–>pdb的预测工具,也是alphafold的简化版
colabfold和alphafold rossetafold的区别?
官方说法:https://new.qq.com/omn/20220531/20220531A01DN000.html
为啥要整一个colabfold?
说白了,alphafold需要大量的计算资源和存储资源,光是依赖的数据库就需要几个T,显卡啥的更是要求比较高,这个门槛显然是比较高的,很多人没办法在本地运行alphafold,这时候就开发了一个colabfold来帮助科研人在本地运行sequence–>pdb的预测;
除此以外,还有一个原因,Google的colab版的alphafold运行起来比较不稳定,而且跑起来比较慢,容易掉线,需要翻墙等等有一系列原因
如何使用colabfold?
colabfold官网:https://colabfold.mmseqs.com/
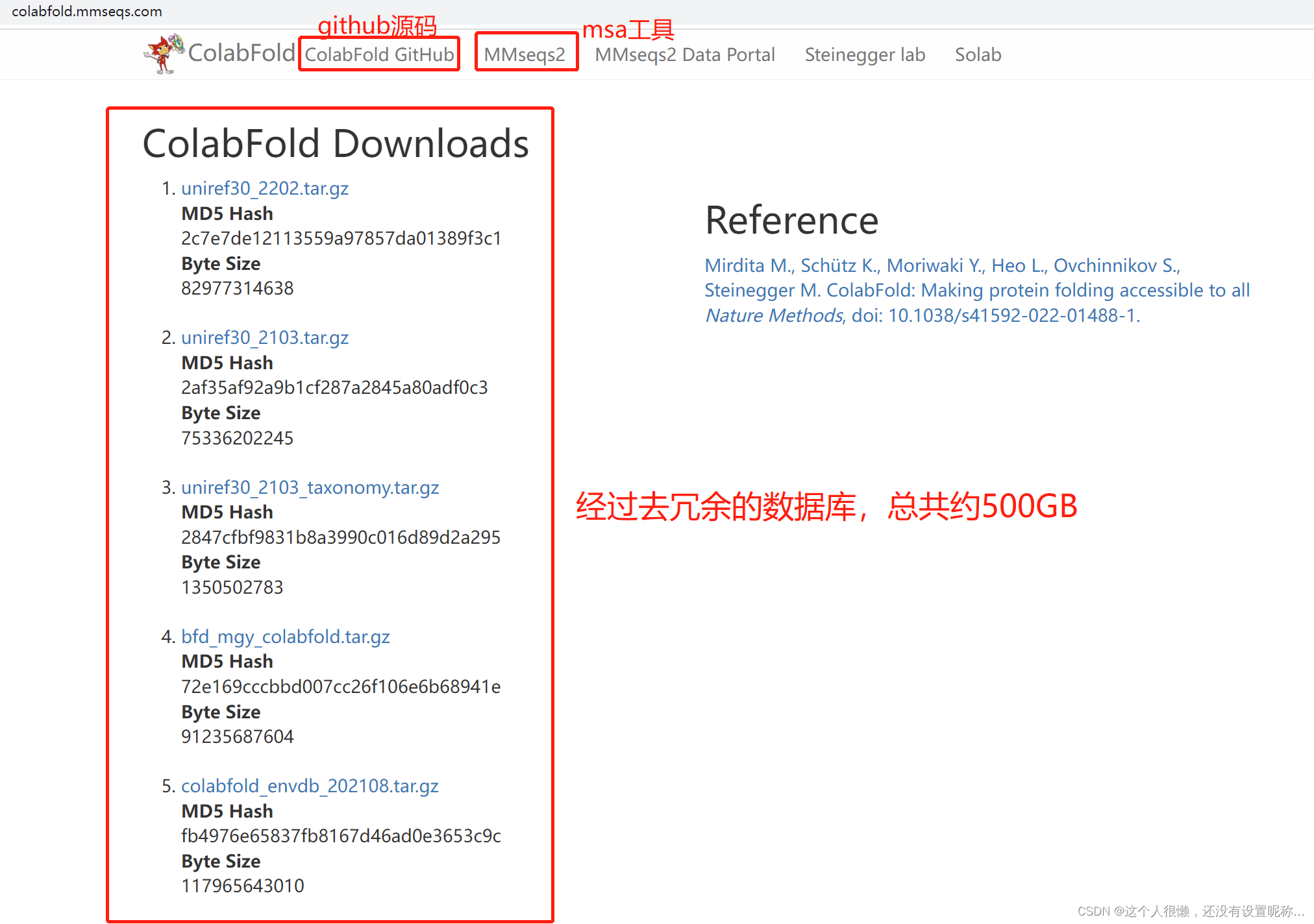 这里的500GB是指压缩包容量,根据readme文件的说法,可能解压后大概是900GB(我猜的),当然,也可以不整数据库,后边说安装的时候就会提到为啥不用整数据库
这里的500GB是指压缩包容量,根据readme文件的说法,可能解压后大概是900GB(我猜的),当然,也可以不整数据库,后边说安装的时候就会提到为啥不用整数据库
colabfold安装?
下载源码:https://github.com/sokrypton/ColabFold
把colabfold的项目源码放到一个固定的位置,然后搭建虚拟环境:
conda create -n colabfold python=3.8
激活环境:
conda activate colabfold
下面的就是readme自带的四条命令,这个按照顺序在虚拟环境中执行就可以
pip install "colabfold[alphafold] @ git+https://github.com/sokrypton/ColabFold"
pip install -q "jax[cuda]>=0.3.8,<0.4" -f https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
# For template-based predictions also install kalign and hhsuite
conda install -c conda-forge -c bioconda kalign2=2.04 hhsuite=3.3.0
# For amber also install openmm and pdbfixer
conda install -c conda-forge openmm=7.5.1 pdbfixer
安装mmseq2:
源码:https://github.com/soedinglab/MMseqs2
安装mmseq2就用下边这条命令
conda install -c conda-forge -c bioconda mmseqs2
至此,环境已经搭好了,下面就可以运行
接下来就是运行了:
colabfold_batch <directory_with_fasta_files> <result_dir>
这条命令就是colabfold给提供的一条,我们可以直接用这条命令直接预测,第一个参数就是fasta路径,第二个就是输出结果的路径。例如:colabfold_batch ./6LO2.fasta ./result 。6LO2.fasta这个文件,可以是单链,可以是双链,双链的话,就是一条一条跑的,最终能跑出来多个结果
如何跑复合物?
例子:XXXX:SSSSSSSSSS,就两条链中间加冒号就可以
mmseq2?
在colabfold的readme中,还有mmseq2这个注释,这个应该是单用的,不需要用在pdb prediction的过程中。我个人认为,如果想用mmseq2这个选项,可以直接按照mmseq2的官网的readme来,但是mmseq2那个官网给的只是单链的alignment,可能paired alignment并不好使;colabfold这个mmseq2是升级版,使用的也是去过冗余的数据库,具体效果我没用过,暂不确定,等用完了再来补;当然,如果只需要完成sequence–>pdb的预测,mmseq2前边的步骤就够用了


















 6409
6409

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











