






声明原文参考:Jim Clauwaert, Gerben Menschaert, Willem Waegeman, DeepRibo: a neural network for precise gene annotation of prokaryotes by combining ribosome profiling signal and binding site patterns,Nucleic Acids Research, Volume 47, Issue 6, 08 April 2019, Page e36,https://doi.org/10.1093/nar/gkz061
代码Github: https://github.com/Biobix/DeepRibo
摘要
由于在不同(亚)物种之间观察到的带注释基因区域的微小变化,原核生物中的基因表达注释经常发现已被纠正。很明显,用于基因组管理的传统序列比对算法无法绘制出完整的基因组图谱。我们提出了DeepRibo,这是一种新颖的神经网络,利用从核糖体谱分析信息中提取的特征和结合位点序列模式显示出了用于描绘和注释原核生物中表达基因的精确工具。该神经网络将循环神经网络和卷积层相结合,将从高通量核糖体谱数据和核糖体结合翻译起始序列区域中获得的信息适配到一个模型中。对翻译领域的先验知识。通过对各种数据集训练的模型,多种物种序列相似性,质谱和经Edman降解验证的蛋白质进行的模型的广泛验证,DeepRibo的有效性得到了强调。
介绍
经过20多年的基因组测序后,很明显细菌中的基因组多样性远远超出了预期,不仅物种之间而且(1)之内。例如,GenBank目前为大肠杆菌(一种原核模式生物之一)拥有超过10,000个基因组装配体,显示出惊人的多样性。跨越所有不同门的大量测序原核生物使得基于序列比对进行基因组比较以阐明基因组复杂性是不切实际的(2)。即使通常使用序列比对,也需要重新注释由于先前注释的基因之间的特性(即DNA序列)的相似性,这些基因是有偏见的,并且已经显示出传播了来自先前错误注释的错误(3)。本文提供的新颖预测工具仅基于从覆盖核糖体结合位点和表达数据的短DNA序列中提取的特征。
开放阅读框(ORF)的划分是在基因注释的基本要素和主要是执行在计算机芯片上(4,5)。核糖体分析(也称为核糖SEQ)测量由测序核糖体保护的片段(与核糖体的mRNA相关的6,7)。核糖SEQ实验使ORF划分,并且该技术已经成功地采用了用于原核生物(8,9)。ORF描绘的一个重要方面是确定翻译起始位点(TIS)。同样,这里还有一些特定的预测工具可以执行此任务(10–12),但这些TIS还可通过在核糖序列协议中添加特定的抗生素治疗(例如氯霉素或四环素)来检测核糖体后进行检测(13)。最近,已经设计了基于机器学习算法的预测方法,以基于核糖体分析和原核基因组序列特征的组合来描绘ORF(14)或预测TIS(15)。真核生物可以使用多种工具(16-21)。
替代proteoform用法也可以通过特定的质谱测量的协议的N-末端肽(研究22,23)。尽管该技术得到认可,但它仍存在缺陷(例如,肽的物理性质和修饰,质谱测量范围……),从而限制了可检测N末端的数量。为了获得更全面的蛋白形式使用图谱,蛋白质组学研究已将上述高通量测序和质谱信息相结合,从而进行了更精确的ORF和TIS验证,从而进行了基因组注释。(24,25)。
在本文中,我们介绍了DeepRibo,这是一种新颖的神经网络实现方法,该方法将核糖体分析数据和结合位点模式应用于原核生物中TIS的精确注释。鉴于已获得足够的数据,已经证明在解决复杂问题方面非常有效的人工神经网络的使用,仍然仅限于生物信息学领域的少数应用。例如,使用卷积神经网络预测与靶蛋白的DNA或RNA结合(26)或调用下一代测序的精确变体(BioRxiv:https ://doi.org/10.1101/092890)。DeepRibo是一个人工神经网络,它同时应用卷积神经网络(CNN)和递归神经网络(RNN)架构,以便处理来自DNA序列和核糖体谱分析信号的信息。神经网络仅处理覆盖核糖体结合区的30个核苷酸的短DNA序列。预测基于从该区域提取的特征,这些特征是通过先验知识选择的,并通过从核糖体特征分析信号提取的特征得到增强。
DeepRibo经过针对各种细菌的可用实验的组合培训,并经过测试可在细菌基因组的从头核糖序列数据中同样出色地工作。我们成功地成功训练了一个高精度模型,该模型能够处理核糖核酸序列数据而不会降低分辨率。我们通过多种物种序列相似性比较(27),可用的质谱数据和翻译起始位点注释(28)进一步验证了我们的结果。
材料和方法
对DeepRibo进行了从核糖体分析数据收集的数据方面的培训。Ribo-seq数据的优点是它不绘制转录的mRNA的未翻译区域。它具有高分辨率和低背景噪音的特点,可以进行精确的基因注释。在原核生物中,没有mRNA的剪接发生,与真核生物相比,沿着编码区域产生了更直接的信号模式。相反,细菌基因紧密堆积且经常重叠,这妨碍了直接注释。为了检测基因组特征,设计该模型以评估一组可能的包含核糖序列信号的ORF,从中选择排名前k位的概率分数来表达基因。此外,该模型在覆盖30nt区域的短DNA序列上进行了训练,该区域与SD主导的基因中的Shine-Dalgarno(SD)基序重叠(在TIS上游最多20nt)和无前导基因中的核糖体结合区(最多TIS下游10nt)。核糖体结合位点已被证明对预测TIS(10,29)。不超过30nt的序列被认为可以阻止DeepRibo进行基因内DNA模式训练。
使用四个参数的S曲线选择样本
使用该物种的最新基因组组装件标记的输入(候选ORF)样本是满足最低信号强度的所有可能ORF的集合。由于核糖体图谱根据实验时生物体的表达情况而变化,因此沿基因组的几个部分不存在信号。实际上,不可能基于表达数据对这些区域进行任何预测。因此,在选择正面和负面数据之前,在训练/评估模型时,不会考虑所有包含低核糖体分析信号的候选ORF。之后,使用从NCBI参考序列(RefSeq)数据库检索的注释对其余数据进行标记。数据的选择基于样本的两个属性,即覆盖范围和信号读取计数。覆盖率指示存在信号的候选ORF的核苷酸部分。信号读取计数,表示为每千千位读取数(RPKM),表示与数据集读取计数相比样品中的读取量。由于考虑的数据集的最大分区覆盖范围从零到低,因此从过滤后的输入样本中可以获得读取计数,覆盖范围和标签值的更平衡的分布。此外,与收集的数据中存在的所有候选ORF相比,最终数据集包含约五分之一的输入样本。由于考虑的数据集的最大分区覆盖范围从零到低,因此从过滤后的输入样本中可以获得读取计数,覆盖范围和标签值的更平衡的分布。此外,与收集的数据中存在的所有候选ORF相比,最终数据集包含约五分之一的输入样本。由于考虑的数据集的最大分区覆盖范围从零到低,因此从过滤后的输入样本中可以获得读取计数,覆盖范围和标签值的更平衡的分布。此外,与收集的数据中存在的所有候选ORF相比,最终数据集包含约五分之一的输入样本。
为了确定覆盖范围和RPKM的最小截止值,Ndah 等人介绍了一种方法。(14)已被应用。该方法基于Lutz 等人完成的阈值剂量反应估算。(30)。为此,在RPKM的功能范围内,安装了四个参数的S曲线。拟合S曲线时仅考虑阳性样本。通过预测拟合的S曲线的下弯曲,可以获得每个数据集的信号覆盖率和RPKM的最小截止值。这一点很重要,因为它可以将可与背景噪声区分开的阳性样本分开。该点被定义为正标记的候选ORF内RPKM的增加与所述数据集中核糖序列信号的覆盖范围相关的点。使用S技术,可以将来自几个单独实验的数据合并起来,因为S曲线分别适合每个数据集。
为了标记样品,使用了提及物种的公共基因组注释。实际上,已经做出了这样的假设:经过序列比对标记的数据训练的DeepRibo可以通过从核糖序列信号中学习而不是使用完整的DNA序列作为输入来提供精确的预测。尽管由于普遍但更保守的DNA序列比对方法的缺点,预计带注释的基因组会包含错误,但是由于模型不学习编码序列的DNA序列,因此无法模仿这种行为。
神经网络架构
DeepRibo是内置在PyTorch(31)中的神经网络,其架构如图1所示 。它是专门为处理两种类型的数据而设计的:字符串(即DNA序列)和浮点型(即ribo-seq信号)。该模型首先并行处理每种类型的数据,然后再将这两个输入创建的特征组合到一组完全连接的层中。DNA序列被转换为具有四个通道的二进制图像,这是Alipanahi(26)。该图像由两个卷积层连续处理。第一层使用四个1×1卷积核将稀疏矩阵转换为密集矩阵。之后,32个1×12卷积的内核在第二个和最后一个卷积层中处理数据。核糖体图谱数据被送入双层双向双向门控复发单元(GRU)。选择门控循环单元代替长期的短期记忆单元,因为它显示出可以训练更好的模型,并且总体上可以更快地训练。仅检索存储单元的最终隐藏状态以进行进一步处理,从而可以使用长度可变的输入(即候选ORF)。在处理每种类型的数据之后,将两个网络的输出节点连接起来并馈入一个完全连接的层中。网络的最终层由三个完全连接的层组成,这些层结合了卷积神经网络(CNN)和递归神经网络(RNN)的功能以获得最终预测。整流后的线性单位用作除最后一层以外的每一层的激活函数。在训练过程中,将二进制交叉熵用作损失函数。

神经网络DeepRibo的体系结构。对于每个候选ORF,将处理两种类型的数据,并将其输入到神经网络的相应部分。卷积层在30个核苷酸的DNA序列上训练,范围从TIS的上游20个核苷酸到下游的10个核苷酸。循环神经网络覆盖了起始密码子上游50个核苷酸(包括SD区域)的完整ORF,并延伸了终止密码子下游20个核苷酸的完整ORF。首先将DNA序列翻译成二进制图像,然后分别由四个1×1和32 1×12卷积核处理。核糖体图谱数据由128个隐藏节点的双层双向GRU处理。
数据集构建
已包括几个用于训练的数据库,包括对在标准条件下生长的原核生物进行的实验。实验涵盖革兰氏阴性(鼠伤寒沙门氏菌(14),大肠杆菌(32),新月形杆菌(33))和革兰氏阳性细菌(枯草芽孢杆菌(34),耻垢分枝杆菌(35),金黄色葡萄球菌(36),天蓝色链霉菌(37))。使用核糖体图谱覆盖信号训练模型。将S曲线拟合到每个数据集上,以获取每个数据集中样本的核糖体分析信号的最小所需覆盖率和RPKM信号。表 1概述了所使用的数据集,每个样本的数量均对正/负数据集有所贡献。

为了确保在输入数据的创建过程中不引入偏差,第一步是为每个包含的核糖序列数据库选择基因组的所有候选ORF。研究表明,ATG,GTG和TTG是三种核苷酸的组合,几乎完全构成了各种细菌中所有起始密码子(38)。因此,在本研究中,从ATG,GTG和TTG到终止密码子(TAA,TGA或TAG)开始的基因组内的所有DNA序列均被视为候选ORF。由于存在大量ORF,它们的长度太短而无法翻译成功能蛋白,因此选择30个核苷酸的伪任意截止点作为样品的最小长度。
该研究的构建如下:从七个可用数据集中的六个中创建六个训练数据,并使用其余数据集作为测试集。使用每个可用数据集作为测试集,总共对七个模型进行了培训和评估,以进行本研究。此外,本研究突出了两种模型的性能。在第一个设置中,我们从训练集中排除金黄色葡萄球菌的数据。在第二个设置中,来自大肠杆菌的数据被排除在训练集中。两种设置都覆盖了RPKM和带注释基因的覆盖范围之间最低和最高相关性的数据集。所有实验均评估了DeepRibo在从头开始的性能数据(即转移学习),根据所讨论的设计目标。由六个数据集构成的训练数据分为训练集(95%)和验证集(5%)。验证集的丢失用于确定训练停止的时间点。补充图S3和S4在所有评估模型的训练,验证和测试集上可视化模型的损失。
评估和后处理
为了评估模型,使用了精确召回曲线下的面积(PR AUC)性能度量。由于实验设置,输入样本的标签高度不平衡。因此,误报率的大变化只会导致误报率的小变化。由于模型的最终用途集中在对排名靠前的k个基因的预测上,因此PR AUC被认为是一种更具信息量的措施(39)。的确,即使在假阳性的绝对数量(严重)超过真实阳性的绝对数量的情况下,测得的接收器工作面积下的特征曲线(ROC AUC)值也可能很高。
模型给出的注释的一个重要的后处理步骤是决定是否可以为每个终止密码子仅注释一个TIS。RNA的翻译速率反映的测序深度在不同基因区域之间差异很大。由于不同的RPKM速率,在基因区域之间存在概率得分分布的差异。因此,发生了在一个区域内注释多个起始位点,而在另一区域内未获得TIS的情况。为了将模型与从NCBI检索的注释(不支持多个起始位点)进行比较,重点仅放在两个终止密码子之间的最高预测概率(单个起始位点设置)上。为了获得一组预测结果,必须设置概率得分的阈值,确定排名最高的k个预测ORF的注释。在这项研究中,为每种生物设定了阈值,以便获得与阳性标记的ORF相等数量的阳性预测。
基于局部比对的多序列比较
给定每种模型的性能指标,就可以对结果进行更深入的探索。假设注释文件中存在不完整和错误,已使用基本局部比对搜索工具(BLAST)对DeepRibo进行的注释与程序集之间的差异进行了调查(27)。将模型的假阳性预测与包含先前在文献中讨论过的蛋白质集合的数据库进行比较,从而形成了评估预测的ORF存在的良好标准。查询有关“非冗余蛋白序列”的假阳性预测(包含来自GenBank翻译的非冗余序列以及Refseq,PDB,SwissProt,PIR和PRF的序列(40))已使用蛋白质BLAST(pBLAST)进行。期望值(E)的最大截止值为0.1 。该Ë值给出命中覆盖给出的数据库的大小类似的对齐的预期量。为了清楚起见,错误的阳性预测被认为是可能的蛋白形式或新蛋白,因此被如此标记。具体而言,与呈阳性标记的ORF相比,蛋白形构成的假阳性预测起始点有所不同。新蛋白涵盖了任何先前没有注释的预测ORF。
结果
截止值的S曲线估计可从低质量数据中过滤出高质量图像
为了归一化多个数据集之间的总信号计数,假设不同实验的表达率相等。由于我们不使用同一实验的重复项,因此在合并数据集之前不会进行任何归一化处理。但是,不同实验之间总体信号强度的差异可能是由于生物表达谱的差异,生长条件的变化或进行研究时引入的技术差异引起的。为了过滤信号强度与背景噪声无法区分的候选ORF,请使用S曲线方法(30)为每个数据集估计最小截止值(补充图S1)。有趣的是,包含大量低表达值的数据集会产生更严格的临界值(例如金黄色葡萄球菌)。在表达基因和未表达基因之间有明显区别的情况下,可获得相对较低的临界值(例如大肠杆菌)。因此,根据数据的质量,从每个数据集中选择的样本数量可能会有很大差异。大肠杆菌和金黄色葡萄球菌数据集的阳性样品和拟合的S曲线绘制在图 2中。在注释数据集不正确的情况下,可以预期正样本的覆盖率和RPKM之间的相关性会降低,并且数据点将向较低的RPKM和覆盖率值转移。由于这些元素使S曲线的较低弯曲点在数据上更渐进地拟合,因此这些估计的截止值将更高。
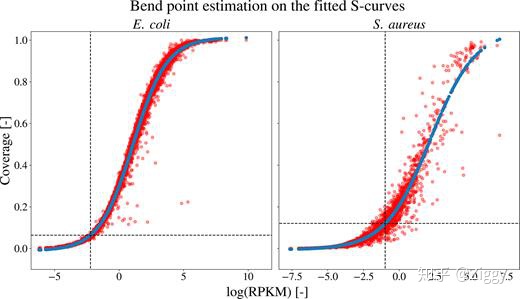
针对大肠杆菌(左)和金黄色葡萄球菌(右)数据集的对数RPKM函数拟合覆盖率的拟合S曲线的折点估计。将每个数据集(红色)的正样本与拟合的S曲线的预测样本(蓝色)作图。对于每个数据集,使用弯曲电缆函数估算拟合曲线的较低弯曲点,以获得最小截止值。
在单个和多个起始密码子的情况下预测的高性能值
为了评估性能,对测试集进行了过滤以排除任何表达率低且带有正标记的数据。由于这些基因没有被表达,具有不存在的或低核糖序列数据的阳性样品被滤出(见表 1)。在选择训练集的同时,已使用拟合的S曲线确定了最小截止值。表 2显示了独立数据集上所有模型的性能。即使DeepRibo在数据集上进行了训练,对于该数据集,在两个终止密码子中最多存在一个正标记的ORF,但这并未反映在模型的预测中。由于每个终止密码子最多使用一个起始密码子对基因组装配进行注释,因此,AUC和PR AUC评分总体上更好,其中每个终止密码子的起始位点最高。在不同的实验设置之间,模型的性能仅略有不同。对于金黄色葡萄球菌和大肠杆菌,分别在测试集上获得的PR AUC分别高达0.965和0.943 。尽管已经证实原核生物中存在多个起始位点(13),由于核糖序列信号的变化,预计预测在不同区域之间的分布会发生偏移。但是,即使考虑允许多个ORF共享一个终止位点的预测,PR AUC分数也高达0.874。
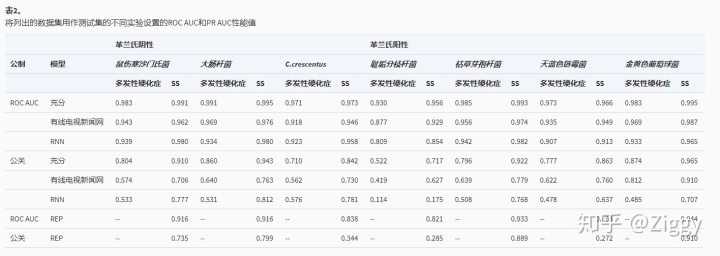
如果认为可能存在多个起始位点(MS),并且每个终止密码子只能具有一个预测的起始位点(SS),则将给出性能指标。如果将两种功能组合在一个模型中(完整),则使用DNA序列作为输入(CNN)或核糖序列数据(RNN)的DeepRibo的性能突出显示了改进的性能。此外,还介绍了REPARATION(REP)的性能。请注意,这些模型都使用交叉验证在列出的数据集中进行了训练和评估。
DeepRibo结合了序列信息和核糖体分析数据
为了确认神经网络将核糖体图谱信号用于其预测的能力,已针对核糖体结合区的序列(基于CNN)或核糖体图谱数据(基于RNN)训练了定制模型。在分别对模型进行DNA序列和核糖核酸序列数据训练的情况下,模型模型的架构保持相似,只是重复或卷积部分丢失。表 2列出了每种设置的两种模型的性能。图 3显示了使用大肠杆菌作为测试集的模型的精确召回曲线。补充图S5给出了其他每个模型的相关图,每个图都显示相似的结果。通过S11。两种方法都证明可以从其特定数据进行训练,对于金黄色葡萄球菌的RNN和CNN的AUC值分别为0.965和0.987 。总体而言,两种架构之间的PR AUC得分显示出CNN的性能优于RNN。与单个部分相比,两个神经网络分区的组合带来了性能上的改进。与CNN相比,PR AUC得分提高了约7%,与RNN相比,PR AUC得分提高了23%,表明该模型能够以有意义的方式组合两种信息。

在新标签页中打开下载幻灯片
大肠杆菌数据集上不同网络的精确调用曲线。如果设置了多个起始站点和单个起始站点,则将给出精确调用曲线。结合了RNN和CNN的完整模型(实线)优于单一的CNN(虚线)和RNN(虚线)架构。
评估无领导者和SD主导的基因
每个门中携带Shine-Dalgarno区的基因比例各不相同。放线菌(S. smegmatis,co.color)的平均含量为19.2%,α-变形杆菌(C. crescentus)的含量为6.3%,γ-变形杆菌(E. coli,鼠伤寒的S. typhimurium)的含量为4.5%,菌丝菌(B. subtilis,S.金黄色葡萄球菌)4.2%无前导基因(41)。与无前导基因不同,SD引导基因由共有序列“ AGGAGG”定义,存在于TIS上游0–20 nt。先前的研究表明无前导基因的TIS下游没有模式(35)。耻垢分枝杆菌和天蓝色链霉菌的整体性能较低提示与被评估生物体基因组中无前导基因的比例相关。相反,没有观察到CNN在放线菌上的表现之间的相关性,显示出与其他装置竞争的结果。然而,在RNN的性能之间观察到性能的高度差异,耻垢分枝杆菌的PR AUC低至0.175 。对ribo-seq数据的调查显示,重复阅读的比例很高(92%),因此在所有使用的数据集中,每个带有正向标记的ORF的唯一阅读计数最低(459.9)。这比低于四倍天蓝色链霉菌(1952.0)和新月柄杆菌(2109.7),并且远低于金黄色葡萄球菌(8114.3),枯草芽孢杆菌(8328.6),鼠伤寒沙门氏菌(23268.5)和大肠杆菌(26908.0)。这些计数与RNN的性能之间的高度相关性突显了高质量数据的重要性。结果,已经评估了脓肿分支杆菌的核糖体图谱(42),以验证DeepRibo在具有更高无领导基因比例的生物体上的适用性。用于评估耻垢分枝杆菌的完整模型和RNN模型的PR AUC分别为0.865和0.577获得的分数与其他生物模型结果相符。在全部七个数据集上进行训练后,使用完整模型的性能会略有提高(PR AUC:0.898)。对于RNN模型(0.569),性能略有降低,表明耻垢分枝杆菌的低质量核糖体分析数据具有负面影响。壳牌等。(35)提出了对耻垢分枝杆菌 150个基因的重新注释。测试集中存在的116个重新注释的ORF中有30个出现在DeepRibo给出的注释中(排名前4607的预测)。
DeepRibo与REPARATION的比较
赔偿(14)是唯一一个对原核生物执行类似任务的工具。但是,REPARATION在某些关键方面采用了不同的方法。通过比较基因组学,使用目标基因组中所有起始密码子ATG,GTG或TTG-的所有候选ORF,通过比较基因组学创建了一个阳性集。负数集是从所有可能的ORF中以起始密码子CTG组合而成的。具体而言,对于每组ORF,共享帧内终止密码子,将采用最长的序列。REPARATION应用随机森林来区分通过比较基因组学(ATG,GTG,TTG)匹配的ORF集合与所有带有起始密码子CTG(负集合)的ORF子集。相比之下,我们方法中的否定集是由所有未由汇编文件明确标记的ORF组成的,正和负集都忽略带有起始密码子CTG的ORF。因此,DeepRibo处理负分数数据的比例更高,在正数集和负数集之间不存在偏差(起始密码子,长度)。因此可以说DeepRibo处理了一个更复杂的问题。DeepRibo在所有七个数据集上均胜过REPARATION(表) 2),与REPARATION相比,显示出更强大的性能。但是,应该在了解两种工具执行不同功能的前提下对比较进行解释。还应该注意的是,通过REPARATION评估的性能也与不同实验的质量相关,在耻垢分枝杆菌,coelicolor和C.crescentus上的性能也得到了改善。出乎意料的低。REPARATION表明,与DeepRibo相比,它对核糖序列数据的质量更为敏感。DeepRibo具有更多优势:(i)输入实验数据的分辨率没有损失;(ii)可以训练单个模型的数据集数量没有限制;(iii)用户可以使用预训练模型。此外,(iv)性能已在独立的测试集上进行了评估(与每个实验使用交叉验证的情况相比)。
埃德曼退化辅助预测的验证
通过使用Edman降解对成熟蛋白质组的N末端残基进行测序,可以验证细胞内某些蛋白质的产生。Ecogene(28)对大肠杆菌 K-12中的922种蛋白质进行了收集,其特征是文献中讨论的所有经过验证的蛋白质。在922种蛋白质中,大肠杆菌中总共表达了838个ORF 。数据集,使用S曲线方法确定。根据先前的方法,使用单个起始站点设置,正面预测由前3544个预测组成。该模型已正确预测了744个(88.8%)基因。23种(2.7%)验证的蛋白质具有不同于注释的TIS,导致815种蛋白质与注释和验证的蛋白质集合相符。没有一个与经验证的蛋白质一致的预测TIS与标记的数据集不一致。注释和Ecogene数据集中存在的815个TIS中有71个(8.7%)未被模型拾取。更重要的是,在排名最高的4400个预测中,实际存在71个虚假阴性中的28个(占39.4%)。由于DeepRibo对新颖的ORF的注释,一些标记为正的输入样本必定会从3544个阳性预测库中排除。这意味着在815个假阴性中,只有43个(5.27%)预测了TIS在标记基因的上下游。
基于N端蛋白质组学的预测验证
除了埃德曼测序(Ecogene数据集)以外,基于质谱的蛋白质组学还可用于验证DeepRibo所做的注释。更具体地说,N末端蛋白质组学是一种使我们能够检测符合引发剂蛋氨酸加工规则的N末端肽的技术。先前已经确定了781个这样的N-末端用于大肠杆菌(14)。表达了与编码序列比对的721个N-末端肽序列,因此存在于测试集中。721个样本中有659个(91.4%)与注释相符。该模型未预测其中的64个(9.7%),其中34个具有不同的TIS,其中30个不在3544个预测中。有趣的是,在表示与RefSeq注释不一致的TIS的62条肽序列中,DeepRibo预测了11条。尽管可以通过核糖体图谱数据表明在与注解不同的位置存在TIS,但这是注解不防水的有形证据,会对模型的性能指标产生负面影响。图 4 给出了带有NCBI注释和预测的两个验证数据集之间的重叠的概述。

在DeepRibo和NCBI RefSeq数据库(标签)提供的注释中,显示通过Edman测序(左)和质谱(右)验证的蛋白质分布的维恩图。分布仅包括使用S曲线方法确定的明确ORF。
假阳性预测的多序列比对
大量的带注释蛋白质存在多种蛋白形式。但是,在基因组组装中仅标注了每种蛋白质的一种。生物变异,生长条件或生长阶段是影响蛋白质表达速率的一些因素。因此,不同实验之间蛋白质表达的多样性产生了注释基因组的变异。已进行了pBLAST搜索以调查假阳性预测是否可能由注释中不存在的表达蛋白形式引起。通过为每个假阳性预测简单地选择最佳比对的蛋白质来创建摘要。已对金黄色葡萄球菌和大肠杆菌的完整假阳性结果进行了pBLAST搜索。将DeepRibo策划的注释与质谱和Edman测序数据集进行比较,得出该模型建议的34和42个具有不同TIS的ORF。此外,还包括了该模型的这两组替代注释,用于序列相似性比较。表 3概述了结果。正如预期的那样,所有蛋白形式均已成功比对,因为它们部分与注释的基因相同。在金黄色葡萄球菌和大肠杆菌中,多达79个中的73(92.4%)和232中的198(85.3%)个带注释蛋白形式。已与数据库中具有共享TIS和终止位点的蛋白质位点完全对齐。在DeepRibo注释的所有新蛋白质中,超过一半的蛋白质具有完全比对的结果,总计对金黄色葡萄球菌和E的25个蛋白质序列(60%)中的15个和258个蛋白质序列(53.1%)中的137个大肠杆菌。有趣的是,相当大比例的新型蛋白质被描述为“假设的”。与MS和Ecogene数据集相比,注释了不同TIS的模型预测大多表明与非冗余数据库中存在的蛋白质完全对齐,在34个匹配项中占28个(82.4%),在43个匹配项中占36个(83.7%),分别。图 5给出了每种比对蛋白的E值分布。补充文件2中提供了有关大肠杆菌和金黄色葡萄球菌的假阳性和假阴性预测的完整列表,包括两个验证集和BLAST结果。
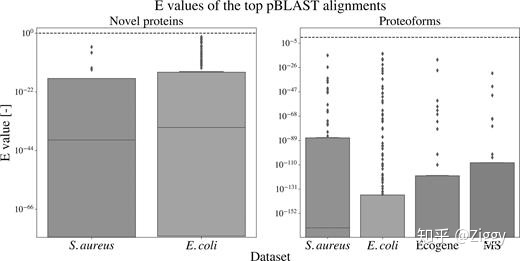
pBLAST的E值分布针对不同数据集的新预测蛋白质(左)和蛋白形式(右)。该ê值给出了最佳命中(如果存在的话),每个误报。虚线表示E值为1。

这些预测可分为蛋白形式和新蛋白,这些蛋白形式的TIS在带注释的ORF的上游或下游,或构成具有无注释的终止位点的ORF。这些蛋白质的BLAST搜索是在非冗余蛋白质数据库上进行的。E分数的最大截止值为0.1。给出每种类型的误报总数。对于每个假阳性结果仅取最佳比对的蛋白质(即最高的E评分),即可给出按起始位点或起始位点和终止位点进行比对的总匹配量。最后,给出了被描述为“假设的”蛋白质的总量。
讨论
在涉及大数据的热门主题上的深度学习方法的成功正在慢慢地进入涉及多组学的生物信息学领域。尽管自第二代测序技术问世以来,就已经可以使用由高通量方法创建的大数据,但到目前为止,主要是使用统计方法来进行探索,但不包括机器学习。事实证明,深度学习非常成功,可以在不希望或无法解释其功能时使用黑盒方法。在这项研究中,我们提供了一个深层神经网络,可使用核糖体分析数据精确注释基因组上表达的蛋白质。该工具使用体内数据表达谱来注释基因组而不使用比较序列比对。DeepRibo使用结合了卷积层和循环存储单元的新型架构,从DNA序列和核糖序列中包含的信息中学习。从机器学习模型获得的结果(在相同的数据集上进行了训练和评估)可能由于过度拟合而高估了它们在新数据上的性能。由于使用了一个在各种现有数据集上训练并在独立测试集上进行评估的单一模型,因此出现了这个问题。此外,在组合的数据集上构建模型会训练模型区分所有数据集上存在的有用特征和与数据集相关的变化,从而无需进行标准化步骤。DeepRibo是第一个在多个数据集上经过训练和验证的原核生物中,精确描绘ORF的工具。此外,它在所有测试的数据集上均优于REPARATION。
在评估DeepRibo的结果时,必须确定某个临界值以从负面指定正面预测。为了评估模型,已将正ORF的数量设置为等于注释中存在的ORF。但是,由于进行了新颖的预测,因此一部分注释样本的排名必然低于前k个预测(特别是在多个起始站点设置中)。此外,还可以通过模型中前k个预测未获得的验证集中的蛋白质分数来反映这一点。没有针对每个实例的最佳截止值,并且必须根据工具的应用来确定,这假定了所需的精度/调用率。
DeepRibo的性能在所有七个测试集中均保持一致,四个数据集的PR AUC得分均> 0.90。在革兰氏阳性菌和革兰氏阴性菌之间没有观察到性能差异。即使相对较低的性能得到返回的耻垢分枝杆菌,为返回的数据集和表演的评价M.脓肿时,另一名成员分枝杆菌家族,与存在的无前导基因的分数无关。取而代之的是,强调了核糖体谱分析实验质量的重要性,每个带有正向标记的ORF的独特读数显示出与各个RNN的性能相关。尽管映射到基因组的绝对读数数目足够,但是重复水平较高,因此独特读数的数目较少,导致核糖体谱分析的分辨率较低。为了保证核糖体谱分析实验的质量,可以使用以下几种工具:FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)用于评估阅读和mQC(https://github.com/ Biobix / mQC),用于评估映射阅读。
由于大多数候选ORF与其他样本共享其终止位点,因此仅在每个组中选择具有最高预测概率的ORF可以始终提供更好的性能。即使在将单个起始站点与多个起始站点设置进行比较时,可以观察到性能有所提高,但后者的性能仍然值得关注。具体而言,在113 228个(89.5%)候选ORF中有105595个与大肠杆菌数据集中的其他样本共享终止位点。一些地区可能有多达一百个TIS。尽管该模型无法处理此信息,但只能对每个样本进行预测,但测试集(MS设置)的PR AUC得分却很高,范围从0.710到0.874(耻垢分枝杆菌除外)。预计该错误的部分原因是由于不同基因组区域之间存在的RPKM值不同。然而,模型的性能表明这种影响是最小的。此外,最近的研究已经发现有多个翻译起始位点(基因13,15,44)。由于注释不支持此功能,因此目前无法在多站点站点设置中正确评估模型。
许多原核系统具有紧密编织的操纵子结构(45),产生可重叠不同目标区域的核糖序列信号。DeepRibo做出的一些错误肯定注释位于这些区域中(图 6D)。RNN处理的核糖体图谱信号周围包含有填充区域,以前可以提高结果性能。
图6。

在新标签页中打开下载幻灯片
DeepRibo示例注释显示在ribo-seq输入信号和RefSeq注释旁边。数据使用GWIPS-viz浏览器(43)格式化并公开托管(请参阅补充数据)。在每个轨道上显示(从上到下):核糖核酸序列信号(有义:橙色,反义:蓝色),测试集中存在的所有ORF样品的TIS,DeepRibo预测的注释与RefSeq组件不一致(预测ORF)和用于标记数据的RefSeq基因组注释(已标记ORF)。(A)对大肠杆菌的最高等级的蛋白形式预测(基因:PqqL,等级:231)。(B)金黄色葡萄球菌的最高等级的蛋白形式预测(基因:UbiE,等级:131)。(C)没有pBLAST比对的大肠杆菌中排名最高的新型蛋白质(等级:1302)。(D)在具有重叠基因的区域中预测的蛋白形式的实例(基因:ybhF,等级:941)。
已知在核糖体谱分析实验中使用抗生素会影响所得信号。在这项研究中,所有实验均采用氯霉素处理,但天蓝色链霉菌(S. coelicolor)除外,后者应用硫链球蛋白。由于天蓝色链霉菌总体得分较低可能与其独特读物计数的降低有关,因此尚不确定硫代链霉菌素的使用是否会影响DeepRibo的性能。尽管这种作用似乎很小,但需要进一步研究不同抗生素治疗对DeepRibo的作用。新的抗生素治疗还可以改善模型的性能。Meydan 等。(46)讨论了使用抗生素retapamulin来增加核糖seq信号分辨率的方法。retapamulin提供的增加的分辨率可能因此改善其结果注释,特别是对于包含重叠基因的区域。
在大肠杆菌模型的情况下,许多新的预测都位于假基因中。通常,在训练/测试样本中不存在与完整假基因区域重叠的候选ORF,因为这些带注释的特征覆盖了具有多个终止密码子的区域。因此,不存在阳性标记的样品。但是,经常在这些位点测量ribo-seq信号,从而为新的(假阳性)预测创建了热点。
该模型对大量新颖的小开放阅读框(sORF)的识别与注释形成了另一种对比。该模型给出的新的ORF预测对大肠杆菌和金黄色葡萄球菌的中位长度为270和63个核苷酸。这些远低于每种物种中注释基因的中位长度(827和723)。ORF的大小会影响通过计算机方法识别sORF的统计方法的功能(47)应用序列比对。假设基因组中存在的sORF数量比注释中当前存在的数量更多。与注释中存在的数量相比,DeepRibo注释的sORF数量更多。具体来说,VanOrsdel等。最近为大肠杆菌提出了32种新的sORF (48)。在数据集中存在的21个sORF中,有5个包含在DeepRibo提供的注释中(顶部3544)。相比之下,RefSeq的注释中实际上只存在一个建议的sORF。在图6C中给出了 用于大肠杆菌的新型sORF的例子。正面注解和新颖预测的ORF长度分布如图1所示。补充图S12。
从pBLAST搜索获得的结果证实了这一点,很可能是,在评估单个起始位点设置的预测时观察到的假阳性分数中有一部分是由于注释未完全映射生物体的翻译复杂性,随后是负面的影响DeepRibo的性能。对于鲜为人知的原核生物,例如天蓝色链霉菌,新月形梭菌,耻垢分枝杆菌和脓肿分枝杆菌,这是特别期望的。在进一步的支持下,对带有核糖核酸序列信号的预测的详细评估表明,通过人工评估可以支持许多假阳性结果(图 6 A / B / C)。
事实证明,DeepRibo是一种具有新颖方法和高性能的工具。该模型能够发现共享一个终止密码子的多个ORF,小的ORF或位于假基因区域的ORF。训练DeepRibo不受任何数量的数据集的束缚,它区分了数据集之间共享的有用功能,并且可以随着添加更多数据而进一步改进。与序列比对方法相比,核糖体图谱信号为ORF的注释提供了实际的证据。从基因中排除DNA序列可确保模型不会偏向于对其进行训练的基因模式。总而言之,DeepRibo已证明是在不使用基因相似性算法的情况下注释基因组的可行工具,并且可用于辅助在原核生物中发现翻译蛋白。
补充数据
可从NAR Online获得补充数据。
致谢
作者感谢根特大学的支持。特别感谢Audrey Mannion-Michel博士在GWIPS-viz方面的工作,并为核糖体分析数据提供了帮助。
资金
根特大学特别研究基金[BOF24j2016001002 to PR];法兰德斯研究基金会(FWO-Vlaanderen)博士后研究金(转给通用汽车)。开放获取费用的资金:Bijzonder Onderzoeksfonds [BOF24j2016001002]。
利益冲突声明。没有声明。
REFERENCES
1. Land M., Hauser L., Jun S.-R., Nookaew I., Leuze M.R., Ahn T.-H., Karpinets T., Lund O., Kora G., Wassenaar T. et al. . Insights from 20 years of bacterial genome sequencing. Funct. Integrative Genomics. 2015; 15:141–161.
Google ScholarCrossref
2. Richardson E.J., Watson M. The automatic annotation of bacterial genomes. Brief. Bioinformatics. 2013; 14:1–12.
Google ScholarCrossrefPubMed
3. Fields A.P., Rodriguez E.H., Jovanovic M., Stern-Ginossar N., Haas B.J., Mertins P., Raychowdhury R., Hacohen N., Carr S.A., Ingolia N.T. et al. . A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol. Cell. 2015; 60:816–827.
Google ScholarCrossrefPubMed
4. Delcher A. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999; 27:4636–4641.
Google ScholarCrossrefPubMed
5. Hyatt D., Chen G.L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119.
Google ScholarCrossrefPubMed
6. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223.
Google ScholarCrossrefPubMed
7. Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802.
Google ScholarCrossrefPubMed
8. O’Connor P.B.F., Li G.W., Weissman J.S., Atkins J.F., Baranov P.V. RRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics. 2013; 29:1488–1491.
Google ScholarCrossrefPubMed
9. Mohammad F., Woolstenhulme C.J., Green R., Buskirk A.R. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 2016; 14:686–694.
Google ScholarCrossrefPubMed
10. Tech M., Morgenstern B., Meinicke P. TICO: a tool for postprocessing the predictions of prokaryotic translation initiation sites. Nucleic Acids Res. 2006; 34:W588–W590.
Google ScholarCrossrefPubMed
11. Ou H.Y., Guo F.B., Zhang C.T. GS-Finder: a program to find bacterial gene start sites with a self-training method. Int. J. Biochem. Cell Biol. 2004; 36:535–544.
Google ScholarCrossrefPubMed
12. Zhu H.-Q., Hu G.-Q., Ouyang Z.-Q., Wang J., She Z.-S. Accuracy improvement for identifying translation initiation sites in microbial genomes. Bioinformatics. 2004; 20:3308–3317.
Google ScholarCrossrefPubMed
13. Nakahigashi K., Takai Y., Kimura M., Abe N., Nakayashiki T., Shiwa Y., Yoshikawa H., Wanner B.L., Ishihama Y., Mori H. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res. 2016; 23:193–201.
Google ScholarCrossrefPubMed
14. Ndah E., Jonckheere V., Giess A., Valen E., Menschaert G., Van Damme P. REPARATION: ribosome profiling assisted (re-)annotation of bacterial genomes. Nucleic Acids Res. 2017; 45:e168.
Google ScholarCrossrefPubMed
15. Giess A., Jonckheere V., Ndah E., Chyzyńska K., Van Damme P., Valen E. Ribosome signatures aid bacterial translation initiation site identification. BMC Biol. 2017; 15:e76.
Google ScholarCrossref
16. Lee S., Liu B., Lee S., Huang S.-X., Shen B., Qian S.-B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432.
Google ScholarCrossrefPubMed
17. Crappé J., Ndah E., Koch A., Steyaert S., Gawron D., De Keulenaer S., De Meester E., De Meyer T., Van Criekinge W., Van Damme P. et al. . PROTEOFORMER: Deep proteome coverage through ribosome profiling and MS integration. Nucleic Acids Res. 2015; 43:e29.
Google ScholarCrossrefPubMed
18. Bazzini A.A., Johnstone T.G., Christiano R., MacKowiak S.D., Obermayer B., Fleming E.S., Vejnar C.E., Lee M.T., Rajewsky N., Walther T.C. et al. . Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014; 33:981–993.
Google ScholarCrossrefPubMed
19. Chew G.-L., Pauli A., Rinn J.L., Regev A., Schier A.F., Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5’ leaders of coding RNAs. Development. 2013; 140:2828–2834.
Google ScholarCrossrefPubMed
20. Xiao Z., Huang R., Xing X., Chen Y., Deng H., Yang X. De novo annotation and characterization of the translatome with ribosome profiling data. Nucleic Acids Res. 2018; 46:e61.
Google ScholarCrossrefPubMed
21. Erhard F., Halenius A., Zimmermann C., L’Hernault A., Kowalewski D.J., Weekes M.P., Stevanovic S., Zimmer R., D”olken L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods. 2018; 15:363–366.
Google ScholarCrossrefPubMed
22. Staes A., Impens F., Damme P.V., Ruttens B., Goethals M., Demol H., Timmerman E., Vandekerckhove J., Gevaert K. Selecting protein n-terminal peptides by combined fractional diagonal chromatography. Nat. Protocols. 2011; 6:1130–1141.
Google ScholarCrossref
23. Berry I.J., Steele J.R., Padula M.P., Djordjevic S.P. The application of terminomics for the identification of protein start sites and proteoforms in bacteria. PROTEOMICS. 2016; 16:257–272.
Google ScholarCrossrefPubMed
24. Hartmann E.M., Armengaud J. N-terminomics and proteogenomics, getting off to a good start. PROTEOMICS. 2014; 14:2637–2646.
Google ScholarCrossrefPubMed
25. Van Damme P., Gawron D., Van Criekinge W., Menschaert G. N-terminal proteomics and ribosome profiling provide a comprehensive view of the alternative translation initiation landscape in mice and men. Mol. Cell. Proteomics. 2014; 13:1245–1261.
Google ScholarCrossrefPubMed
26. Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838.
Google ScholarCrossrefPubMed
27. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410.
Google ScholarCrossrefPubMed
28. Zhou J., Rudd K.E. EcoGene 3.0. Nucleic Acids Res. 2013; 41:D613–D624.
Google ScholarCrossrefPubMed
29. Zhu H., Hu G.-Q., Yang Y.-F., Wang J., She Z.-S. MED: a new non-supervised gene prediction algorithm for bacterial and archaeal genomes. BMC Bioinformatics. 2007; 8:97.
Google ScholarCrossrefPubMed
30. Lutz R.W., Stahel W.A., Lutz W.K. Statistical procedures to test for linearity and estimate threshold doses for tumor induction with nonlinear dose-response relationships in bioassays for carcinogenicity. Regul. Toxicol. Pharmacol. 2002; 36:331–337.
Google ScholarCrossrefPubMed
31. Paszke A., Gross S., Chintala S., Chanan G., Yang E., DeVito Z., Lin Z., Desmaison A., Antiga L., Lerer A. Automatic differentiation in PyTorch. NIPS-W. 2017;
Google Scholar
32. Li G.W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635.
Google ScholarCrossrefPubMed
33. Schrader J.M., Zhou B., Li G.W., Lasker K., Childers W.S., Williams B., Long T., Crosson S., McAdams H.H., Weissman J.S. et al. . The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014; 10:e1004463.
Google ScholarCrossrefPubMed
34. Li G.W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012; 484:538–541.
Google ScholarCrossrefPubMed
35. Shell S.S., Wang J., Lapierre P., Mir M., Chase M.R., Pyle M.M., Gawande R., Ahmad R., Sarracino D.A., Ioerger T.R. et al. . Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLOS Genet. 2015; 11:e1005641.
Google ScholarCrossrefPubMed
36. Davis A.R., Gohara D.W., Yap M.-N.F. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15379–15384.
Google ScholarCrossrefPubMed
37. Jeong Y., Kim J.N., Kim M.W., Bucca G., Cho S., Yoon Y.J., Kim B.G., Roe J.H., Kim S.C., Smith C.P. et al. . The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat. Commun. 2016; 7:11605.
Google ScholarCrossrefPubMed
38. Panicker I.S., Browning G.F., Markham P.F. The effect of an alternate start codon on heterologous expression of a PhoA fusion protein in mycoplasma gallisepticum. PLoS ONE. 2015; 10:e0127911.
Google ScholarCrossrefPubMed
39. Davis J., Goadrich M. The relationship between precision-recall and ROC curves. Proceedings of the 23rd International Conference on Machine Learning. 2006; NYACM ICML ’06233–240.
Google Scholar
40. Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007; 35(Suppl. 1):D61–D65.
Google ScholarCrossrefPubMed
41. Zheng X., Hu G.-Q., She Z.-S., Zhu H. Leaderless genes in bacteria: clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genomics. 2011; 12:361.
Google ScholarCrossrefPubMed
42. Miranda-CasoLuengo A.A., Staunton P.M., Dinan A.M., Lohan A.J., Loftus B.J. Functional characterization of the Mycobacterium abscessus genome coupled with condition specific transcriptomics reveals conserved molecular strategies for host adaptation and persistence. BMC Genomics. 2016; 17:553.
Google ScholarCrossrefPubMed
43. Michel A.M., Fox G.M., Kiran A., De Bo C., O’Connor P.B., Heaphy S.M., Mullan J.P., Donohue C.A., Higgins D.G., Baranov P.V. GWIPS-viz: Development of a ribo-seq genome browser. Nucleic Acids Res. 2014; 42:D859–D864.
Google ScholarCrossrefPubMed
44. Dai Y., Shortreed M.R., Scalf M., Frey B.L., Cesnik A.J., Solntsev S., Schaffer L.V., Smith L.M. Elucidating Escherichia coli proteoform families using intact-mass proteomics and a global PTM discovery database. J. Proteome Res. 2017; 16:4156–4165.
Google ScholarCrossrefPubMed
45. Pallejà A., Harrington E.D., Bork P. Large gene overlaps in prokaryotic genomes: result of functional constraints or mispredictions?. BMC Genomics. 2008; 9:335.
Google ScholarCrossrefPubMed
46. Meydan S., Vázquez-Laslop N., Mankin A.S. Genes within genes in bacterial genomes. Microbiol. Spectrum. 2018; 6:doi:10.1128/microbiolspec.RWR-0020-2018.
47. Pauli A., Valen E., Schier A.F. Identifying (non-)coding RNAs and small peptides: Challenges and opportunities. BioEssays. 2015; 37:103–112.
Google ScholarCrossrefPubMed
48. VanOrsdel C.E., Kelly J.P., Burke B.N., Lein C.D., Oufiero C.E., Sanchez J.F., Wimmers L.E., Hearn D.J., Abuikhdair F.J., Barnhart K.R. et al. . Identifying new small proteins in Escherichia coli. Prpteomics. 2018; 18:1700064.
Google ScholarCrossref















 1304
1304











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









