







NGSCheckMate:数据配对检查好工具
求知若渴 没有BUG
背景
生信小灶
肿瘤NGS或者其它类似数据分析工作,大的前提就是样本使用的正确性,然而很多意外的情况都会导致样本的错误使用,对它们进行质控显得尤其重要。
原理
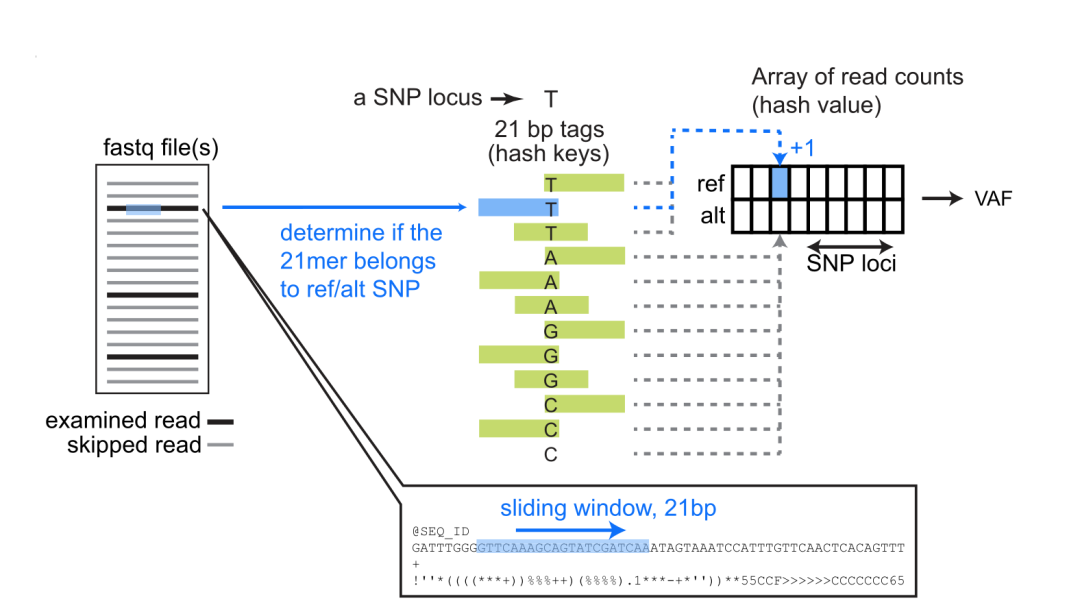
软件处理fasq、bam、vcf等格式,获得VAF的信息,通过计算VAF的相关性,来判断样本是否来自同一个样本。

如何实现fastq的VAF统计?
vcf文件中直接存在有VAF的信息,对于bam,可以转化成mpileup格式获得VAF信息,而对于未经比对的fastq,获得VAF是一件比较困难的事情,作者的处理非常巧妙:软件预构建了一个参考SNP的21-kmer的哈希表,通过reads提取的21-mer与哈希表匹配计算count,获得VAF的参考信息。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 3292
3292











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









