







欢迎大家关注 微信公众号|计算生物前沿
浙江大学团队开发了一款名为Delete的AI药物设计模型,通过结合生成式人工智能与结构生物学方法,实现了针对蛋白口袋的先导化合物“一站式”优化。该模型在保留核心药效团的同时,能精准优化分子结合力与成药性,并在针对LTK蛋白的抑制剂设计中成功获得纳摩尔级活性分子(IC₅₀=1.36 nM),为快速可控的理性药物设计提供了新工具。

引言
当前挑战:首先是数据依赖性强,传统AI药物设计依赖大量已知活性分子数据,难以适用于新靶点。其次是当前模型忽视三维结构,多数模型仅生成二维分子结构,忽略蛋白-配体空间相互作用。此外现有工具功能单一,现有工具仅能完成“分子生成”“连接链设计”等单一任务,缺乏整合性。
Delete的解决方案:该模型具有空间感知能力,它基于等变网络(Equivariant Network)解析蛋白口袋的几何与能量特征。具有多任务兼容特性,通过统一掩蔽策略(Unified Masking Strategy)支持片段扩展、连接链设计、骨架跃迁等四大优化任务。该方法无需预训练数据,通过物理约束而非数据记忆,适配未知靶点的药物设计。
Delete方法概述
Delete是一种“口袋感知”的三维分子生成模型,专注于先导化合物优化。其核心是通过结构生物学数据驱动,将蛋白结合口袋的几何与能量特征融入分子设计,突破传统方法对二维结构或大量活性数据的依赖,实现更精准的靶向药物设计。
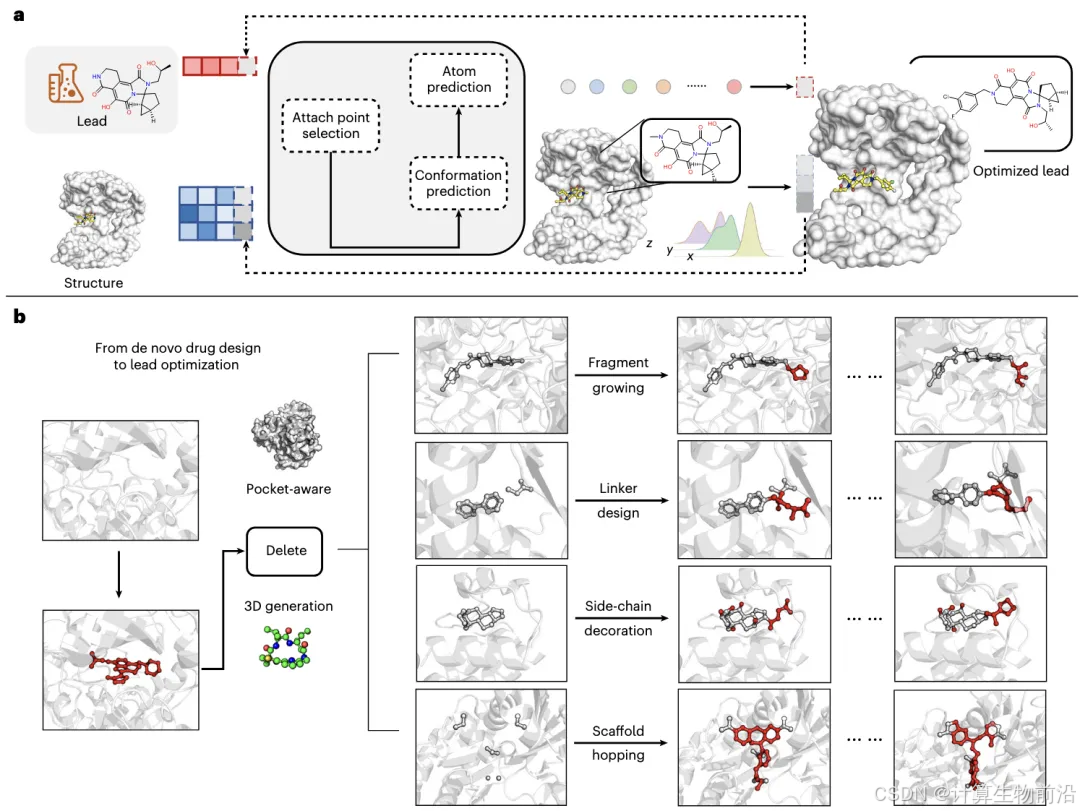
关键技术架构
等变几何网络:解析蛋白口袋的原子坐标与空间取向,确保生成的分子构象满足旋转平移不变性(即无论蛋白结构如何旋转,输出分子拓扑不变);避免数据增强需求,直接生成与口袋匹配的天然构象。
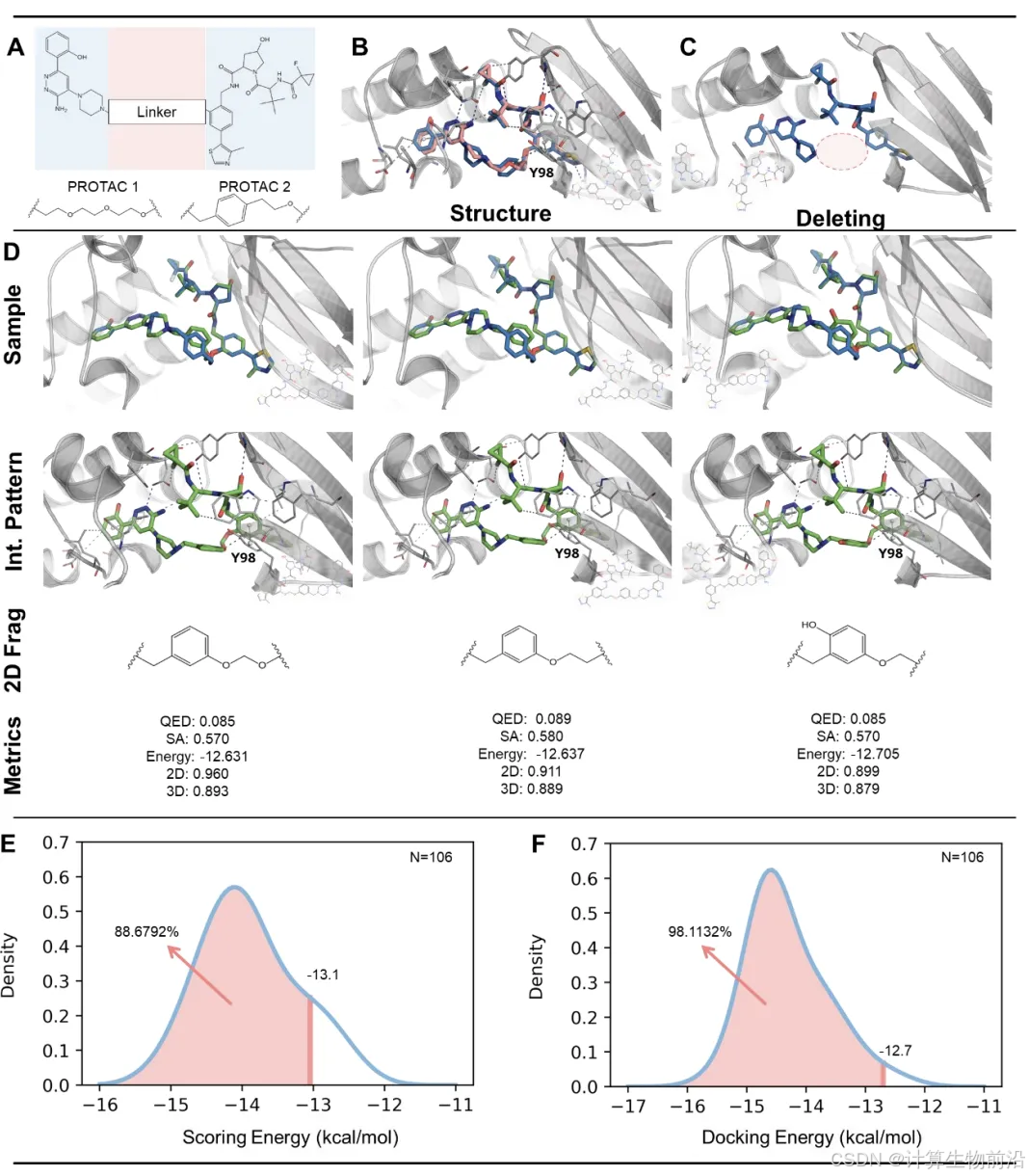
统一掩码策略:通过逐步“删除-预测”循环,动态优化分子结构。例如,在骨架跃迁任务中,先掩蔽原分子骨架,基于保留的侧链碎片生成新骨架。
多任务框架:包括片段扩展、连接链设计、骨架跃迁、侧链修饰;通过蛋白口袋与分子碎片的几何图(Geometric Graph)实时交互,预测原子坐标与键合方式。
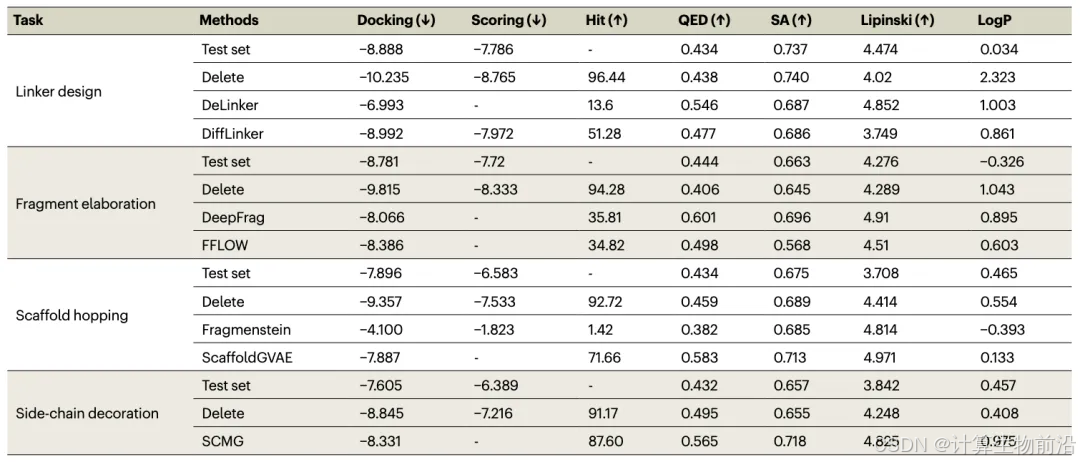
模型性能全面领先
Delete在四大优化任务中结合能(Docking Score)显著优于基线模型。例如,在连接链设计任务中,Delete的结合能(-10.235 kcal/mol)比第二名DiffLinker(-8.992 kcal/mol)提升14%,且93.4%的生成分子满足成药性指标(QED>0.4,SA>0.6)。分子性质(LogP、类药性)与测试集分布高度一致,证明其生成分子兼具高活性与合理性。
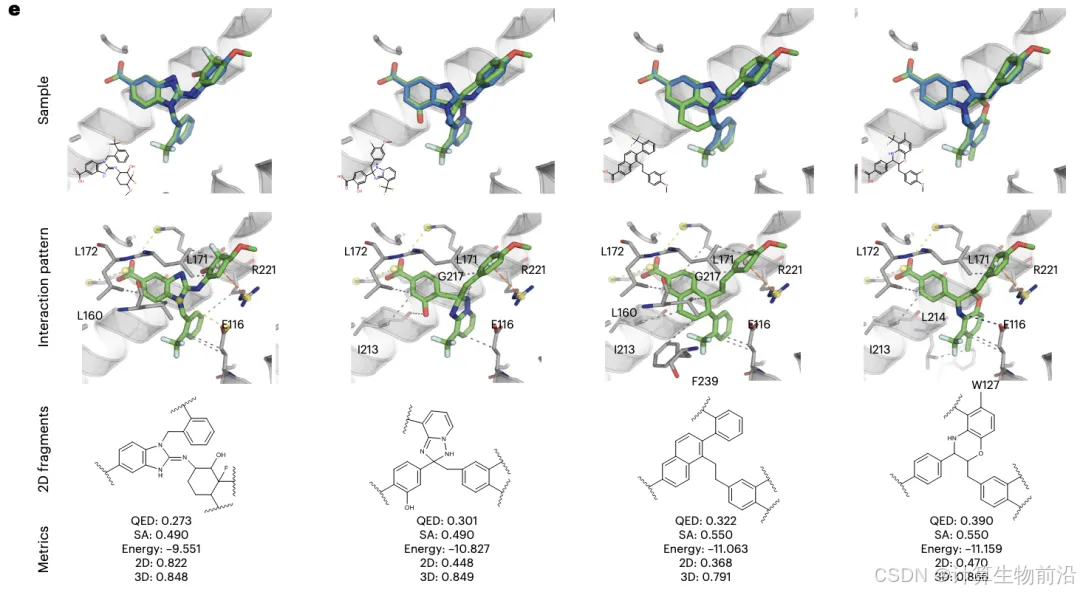
历史案例精准复现
在Eg5蛋白抑制剂骨架跃迁任务中,Delete仅基于原药BI8的碎片(TFs),生成131个新分子,其中40.5%的分子结合能优于原药,且3D构象相似度达0.7878。生成的分子不仅保留关键相互作用(如与残基L172、E116的氢键),还探索了新的疏水相互作用(如与W127、F239)。
LTK抑制剂成功设计
基于ALK抑制剂化学空间迁移学习,生成227个候选分子,其中13个进入实验验证。
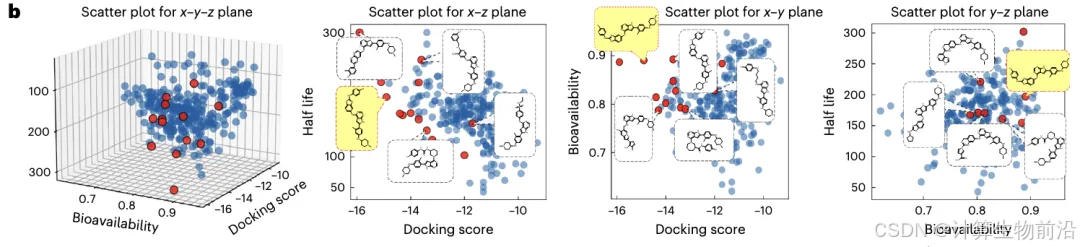
化合物CA-B-1对LTK的IC₅₀达1.36 nM,且选择性抑制31种激酶中的LTK。口服给药(20 mg/kg)显著抑制肿瘤生长,半衰期8小时,药代性质优异。
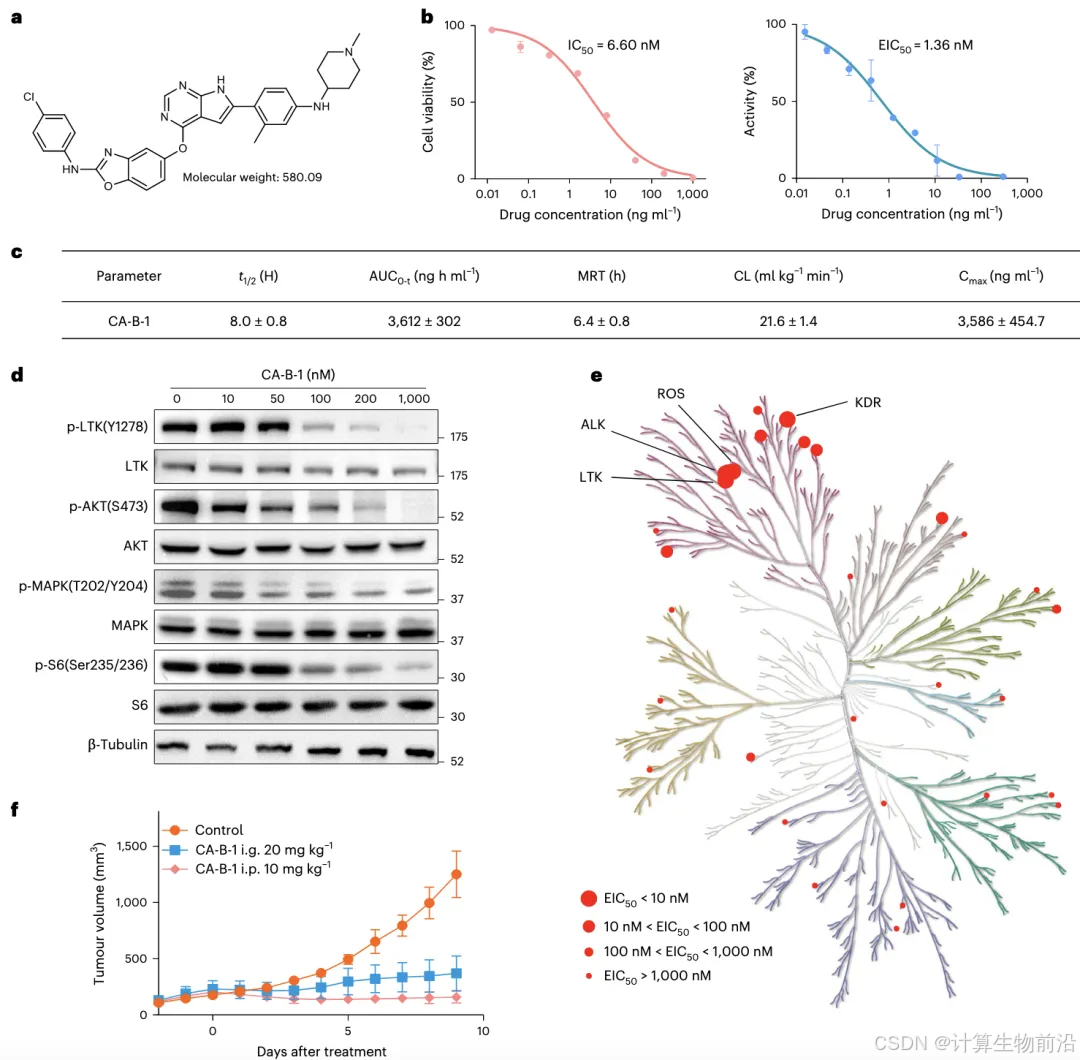
构象质量接近天然
生成分子与蛋白口袋的RMSD(均方根偏差)≤1.2 Å,关键药效团(如氢键、疏水簇)与晶体结构高度一致,验证了Delete在三维空间中的精准建模能力。
参考资料
Chen, S., Zhang, O., Jiang, C. et al. Deep lead optimization enveloped in protein pocket and its application in designing potent and selective ligands targeting LTK protein. Nat Mach Intell (2025). https://doi.org/10.1038/s42256-025-00997-w

















 1696
1696

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









