







基础知识
SNP位点与等位基因的关系
- SNP位点:
SNP(Single Nucleotide Polymorphism,单核苷酸多态性)是指在基因组中某个特定位置上发生的单个核苷酸(A、T、C、G)的变异。 - 等位基因:
等位基因(Allele)是指在同一基因位点上存在的不同变异形式。
对于SNP位点,等位基因就是该位置上可能出现的不同核苷酸(如A或T)。
例如,在基因组位置1000处,某个个体可能是A,而另一个个体可能是T。
SNP的唯一标识
SNP(Single Nucleotide Polymorphism,单核苷酸多态性) 在基因组中的唯一标识,主要包括以下几种形式:
- 基因组坐标(Chromosome Position)
每个 SNP 在基因组中都有一个唯一的位置,通常由 染色体号 + 坐标位置 组成。例如:
chr1:123456
chr2:789012
这种格式是 SNP 的物理位置信息,通常用于基因组参考数据库(如 dbSNP、1000 Genomes)中。
- rsID(Reference SNP Cluster ID)
rsID 是 dbSNP 数据库为每个已知 SNP 分配的唯一标识符,例如:
rs12345
rs67890
如果一个 SNP 已被记录在 dbSNP 中,它会有一个 rs 编号,这是最常见的 SNP 唯一标识方式。
水稻基因编号
水稻的基因号大致分为两类,
一类是RAP号,格式为“Os-Chr-g-number”,
另一类是MSU号,格式为“LOC_Os-Chr-g-number”
常用的水稻基因组数据库
- RAP-db
https://rapdb.dna.affrc.go.jp/ - MSU-RGAP
http://rice.uga.edu/index.shtml
GWAS相关原理
GLM模型
广义线性模型(GLM, Generalized Linear Model)是关联分析中常用的一种统计模型,用于研究基因型与表型之间的关系。
通俗讲
搞统计的经常遇到已知两个变量可能存在一些关系,
使用线性回归模型建模并分析相关系数R和显著性P, 来确定两个变量的相关程度。
同样的,在基因数据的关联研究中,
表型数据与某一SNP位点转化为的数值可以看做两个变量 (数据长度就是样本数)
使用线性回归模型建模并分析相关系数R和显著性P,来确定两个变量的相关程度。
当你把百万级别的SNP位点全部计算完, 再根据一定的阈值去筛选显著性P, 来识别显著的SNP位点。
理论上在R语言能够自己写代码实现GWAS的GLM模型计算,只不过过于费时间了
mod_snp1 = lm(phenotype ~ snp1, data=data_merge) # data_merge为合并的表型与基因型数据
summary(mod_snp1) # 查看显著性P
基本原理图:

SNP位点转换为数值
二倍体
SNP位点数据通常是分类数据(如AA、AT、TT),需要转换为数值形式才能参与计算。常见的转换方法包括:
- 加性模型(Additive Model)
将SNP位点转换为0、1、2,表示次等位基因的拷贝数。
AA → 0; AT → 1; TT → 2 - 显性模型(Dominant Model)
将SNP位点转换为0或1,表示是否携带次等位基因。
AA → 0; AT/TT → 1 - 隐性模型(Recessive Model)
将SNP位点转换为0或1,表示是否纯合携带次等位基因。
AA/AT → 0; TT → 1 - 基因型编码(Genotype Coding)
使用哑变量(Dummy Variable)编码,将SNP位点转换为多个二元变量。
AA → (0, 0); AT → (1, 0); TT → (0, 1)
单倍体
单倍体SNP位点数据通常以单个等位基因的形式存在,需要转换为适合分析的数值格式。以下是常见的转换方法:
- 二元编码
方法:将每个SNP位点的等位基因转换为0或1。
A → 0; T → 1 - 频率编码
方法:根据等位基因在群体中的频率进行编码。
次等位基因频率(MAF)低于0.5的等位基因编码为1,否则为0。 - 矩阵格式
方法:将单倍体SNP位点数据转换为基因型矩阵,行表示个体,列表示SNP位点。
个体1:A T A → 0 1 0; 个体2:T T A → 1 1 0 - 缺失值处理
方法:用特定值(如-9或NA)标记缺失值。
模型公式:

模型拟合
-
最大似然估计(MLE)
-
原理:
通过最大化似然函数寻找最优参数估计。 -
实现方法:
牛顿-拉夫森迭代法(Newton-Raphson)
Fisher评分算法
迭代加权最小二乘法(IRLS)(常规数据集一般就用这个方法,计算稳定,收敛快)
结果解释
系数估计:解释SNP位点对表型的影响。
统计检验:使用Wald检验或似然比检验评估SNP位点的显著性。
p值校正:GWAS中需对p值进行多重检验校正(如Bonferroni校正)
QQ图
此处使用Tassel的GWAS结果绘制QQ图分析。
勾选GLM第一个结果 – Results – QQ Plot
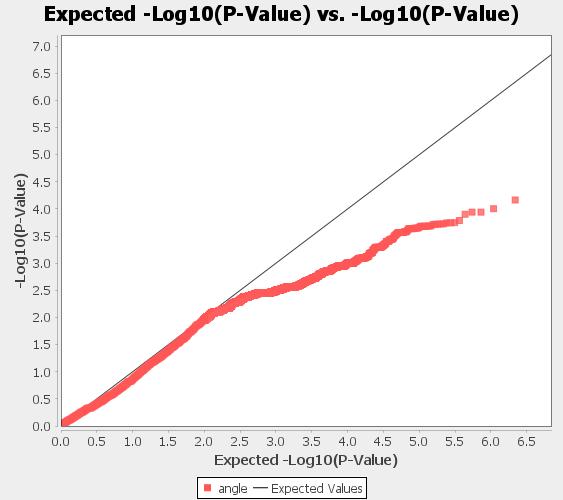
注意上面的图呈现的效果不好,好图是后半段上翘,且尾端有离散点的。
解读QQ图
用于检查系统性偏差
X 轴:理论上期望的 -log10(P-value),假设 P 值符合 均匀分布(无真正的基因关联)。
Y 轴:实际观测到的 -log10(P-value)。
对角线(y=x):表示期望与观察值相符,即 无系统性偏差。
-
理想情况(无系统性偏差)
如果数据点 大部分落在对角线上,说明 GWAS 结果符合期望分布,没有明显的假阳性或系统性偏差。 -
偏离情况 1:数据点整体高于对角线(过度通胀)
可能的原因:
种群结构(Population Stratification) → 需要 PCA 校正
亲缘关系影响(Relatedness) → 需要使用混合线性模型(MLM)
多重检验问题(Inflated False Positives) -
偏离情况 2:数据点整体低于对角线(过度保守)
可能的原因:
校正过度(Overcorrection),如使用了过多的共变量
样本量过小,导致统计效能不足
错误的 P 值计算方法
解决方案:
减少不必要的共变量
增加样本量
检查 GWAS 方法是否合适 -
偏离情况 3:尾部 SNP 明显偏离
只有极端显著的 SNP 偏离(高于对角线),这通常表示 真正的遗传关联信号。
这些点对应 基因组中的重要关联位点,应进一步验证.
曼哈顿图
曼哈顿图
此处使用Tassel软件对GWAS输出结果进行绘图分析。
勾选GLM第一个结果 – Results – Manhattan Plot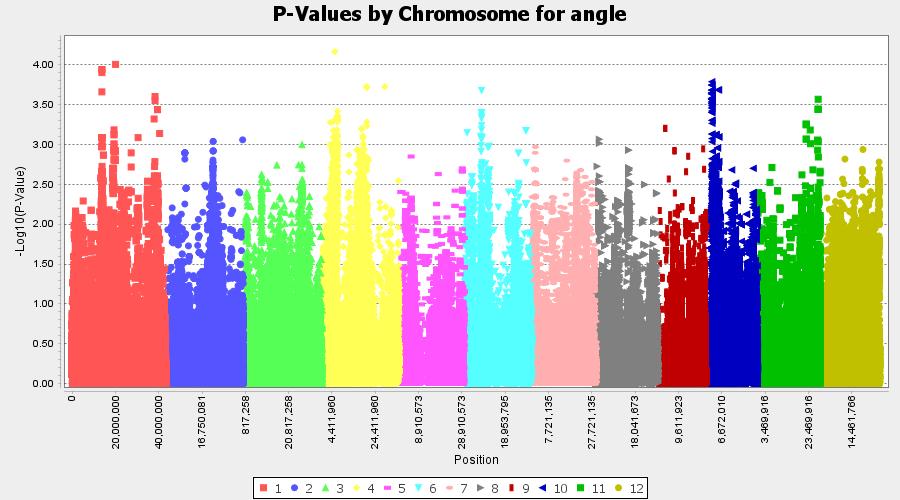
解读曼哈顿图
X 轴(CHR):染色体编号(1-22,X、Y)。
Y 轴(-log₁₀(P-value)):P 值越小(越显著),点的位置越高.
- 显著性阈值(红线):
通常 5 × 10⁻⁸(即 -log₁₀(5e-8) ≈ 7.3)被认为是全基因组水平显著性阈值。
低于 5 × 10⁻⁸ ( -log₁₀(5e-8)大于7.3 )的点一般被认为具有显著的遗传关联。
显著的SNP位点的阈值
一、通用显著性阈值
- 全基因组显著性阈值(Gold Standard)
Bonferroni 校正:
在高通量研究中,常用 Bonferroni 校正 来控制多个比较带来的假阳性风险,计算方式是:
P
<
α
/
S
N
P
数量
P <α / SNP 数量
P<α/SNP数量
其中,α 通常为 0.05,SNP 数量是你测试的 SNP 总数。
如果有 1,000,000 个 SNP,那么 P 值阈值将是:
P
<
0.05
/
1
,
000
,
000
=
5
×
1
0
−
8
P < 0.05 / 1,000,000 = 5 × 10^{-8}
P<0.05/1,000,000=5×10−8
因此 P < 5e-8 是一个常用的显著性阈值。
绘图时,会使用-log₁₀(5e-8) 表示,不然数值太小了。
通常 5 × 10⁻⁸(即 -log₁₀(5e-8) ≈ 7.3)被认为是全基因组水平显著性阈值。
低于 5 × 10⁻⁸ ( -log₁₀(5e-8)大于7.3 )的点一般被认为具有显著的遗传关联。
- 宽松阈值(候选位点筛选)
P < 1×10⁻⁵
适用于探索性分析或样本量较小的研究(如稀有变异分析
二、需调整阈值的情况
- 样本量与人群结构
小样本(N<1,000)或复杂性状:可放宽至P < 1×10⁻⁶,但需后续验证。
超大规模队列(如UK Biobank):可能采用更严格阈值(如P < 1×10⁻⁹)。 - 多重检验校正方法差异
FDR校正:如Benjamini-Hochberg法,常用FDR < 0.05。
置换检验(Permutation):通过数据重抽样确定样本特异性阈值。
注释
GWAS分析找到显著性SNP后,需要注释,才能找到候选的基因。
使用Bedtools注释SNP
- 在ubuntu上安装:
conda install -c bioconda bedtools
bedtools --version
# 下载解压安装
wget https://github.com/arq5x/bedtools2/releases/download/v2.31.1/bedtools-2.31.1.tar.gz
tar zxvf bedtools-2.31.1.tar.gz
cd bedtools2
make
# 测试
bedtools --version
# 使用 head 查看数据
head GWAS.bed
# 使用 bedtools intersect 寻找重叠区域
# -sorted 排序 sorted后的数据运行的更快
# -wa -wb 看具体重叠双方的区域
# > intersect.txt 保存结果
bedtools intersect -a locus.gff -b GWAS.bed -wa -wb > intersect.txt
# 或者不输出,使用 | head -5 查看
bedtools intersect -a locus.gff -b GWAS.bed -wa -wb | head -5
# 使用bedtools merge寻找重叠区域
bedtools merge -i GWAS.bed count > GWAS_merge.bed
# -c 1 -o count 按照第一列计算合并数量
bedtools merge -i GWAS.bed -c 1 -o count > GWAS_merge.bed
# merge要求输入按染色体排序,然后按开始排序坐标。例如,对于 BED 文件,首先对输入进行排序如下:
# sort -k1,1 -k2,2n input.bed > input.sorted.bed
-
学习链接
-
软件github地址:
https://github.com/arq5x/bedtools2/ -
Bedtools官网:
https://bedtools.readthedocs.io/en/latest/ -
官网教程:
http://quinlanlab.org/tutorials/bedtools.html -
官网示例用法:
https://bedtools.readthedocs.io/en/latest/content/example-usage.html -
和刘小泽一起跟着官网学bedtools(官网教程的翻译)
https://www.jieandze1314.com/post/cnposts/151/ -
最全Bedtools使用说明–只看本文就够了
https://www.jianshu.com/p/f8bbd51b5199 -
上海交大超算平台用户手册 Documentation
https://docs.hpc.sjtu.edu.cn/app/bioinformatics/bedtools2.html
注意事项:
-
需要安装在UNIX上
-
Please use tab-delimited BED, GFF, or VCF.
-
官网提示:
从版本 2.28.0 开始,bedtools 现在通过使用 htslib 支持 CRAM 格式。
通过 CRAM_REFERENCE 环境变量指定与您的 CRAM 文件关联的参考基因组。
当 Bedtools 需要从 CRAM 文件(例如 bamtofastq)访问序列数据时,它将查找此环境变量。
除 BAM 文件外,bedtools 假定所有输入文件都以 TAB 键分隔。
bedtools 还假定所有输入文件都使用 UNIX 行尾。
除非使用== -sorted 选项==,否则 bedtools 目前不支持大于 512Mb 的染色体。
当对染色体未按字典排序的文件使用 -sorted 选项时(例如,对 BED 文件使用 -k1,1 -k2,2n 排序),必须提供定义预期染色体顺序的基因组文件 (-g)。
Bedtools 要求您正在比较的文件中的染色体命名方案相同(例如,一个文件中的“chr1”和另一个文件中的“1”不起作用)。
.fai 文件可用作基因组 (-g) 文件。
绘图
Venn图
使用R语言的VennDiagram包 (最多5组)
ggvenn包 (2-4组)
ggVennDiagram包 (显示热力图,颜色深浅表示数量多少)
venn包 (2-7组)
-
参考链接:我汇总了韦恩图(Venn Diagram)所有绘制方法,推荐收藏~~
https://cloud.tencent.com/developer/article/1916088 -
参考链接:【R语言】——VennDiagram包绘制维恩图(保姆级教程)
https://blog.csdn.net/weixin_54004950/article/details/128336522
数据转置,如果不转后头函数venn.diagram对矩阵数值不识别 -
参考链接: 绘制Venv图的网站
https://bioinformatics.psb.ugent.be/webtools/Venn/
https://www.bioladder.cn/web/#/chart/17 -
参考链接:R语言:VennDiagram绘制venn图
https://www.jianshu.com/p/f858521828a5
library(RColorBrewer)
color <- brewer.pal(3, "Set3")
# 个性化参数调整
venn.diagram(
x = list(set1, set2, set3),
category.names = c("Set 1" , "Set 2 " , "Set 3"),
filename = 'venn2.png',
output=TRUE,
# 输出
imagetype="png" , # 类型(tiff png svg)
#height = 1000 , # 高度
#width = 1000 , # 宽度
resolution = 400, # 分辨率
compression = "lzw", # 压缩算法
# 圈
lwd = 5, # 圈线条粗细 1 2 3 4 5
lty = 1, # 线条类型, 1 实线, 2 虚线, blank 无线条
fill = color, # 填充色
col = c("red", 'green', 'blue'), # 线条色
# 数字 number
cex = 2, # 数字大小
fontface = "bold", # 加粗
fontfamily = "sans", # 字体
# 标签 category
cat.cex = 2, # 字体大小
cat.col = c("red", 'green', 'blue'), # 字体色
cat.fontface = "bold", # 加粗
cat.default.pos = "outer", # 位置, outer 内 text 外
cat.pos = c(-27, 27, 135), # 位置,用圆的度数
cat.dist = c(0.055, 0.055, 0.085), # 位置,离圆的距离
cat.fontfamily = "sans", # 字体
rotation = 1 # 1 2 3 旋转确定大打头数据集
)
R语言|CMplot包绘制环形曼哈顿图
https://zhuanlan.zhihu.com/p/519195174
使用于R语言的CMplot包
其他教程
TASSEL亲缘关系分析(视频)
https://www.bilibili.com/video/BV1jM411E76J/?spm_id_from=333.999.0.0&vd_source=c897d3fc116908de1c7dfa6218d4d70f
利用Structure软件进行群体结构分析(视频)
https://www.bilibili.com/video/BV1314y1F7cQ/?spm_id_from=333.999.0.0&vd_source=c897d3fc116908de1c7dfa6218d4d70f
GWAS | 2.群体结构 structure
- https://www.jianshu.com/p/3cb1ff5bf921
基因型数据绘制PCA图和聚类分析图
- https://zhuanlan.zhihu.com/p/434317810
GWAS分析中如何确定显著性阈值
- https://zhuanlan.zhihu.com/p/418267913

安装vcftools
- linus版本下载
conda install -c bioconda vcftools
- vcftools如何在Linux系统中安装
https://blog.csdn.net/yijiaobani/article/details/123092409
SNP位点ID添加
- https://blog.csdn.net/qq_53403084/article/details/135443884
如何在 Ubuntu 20.04 上安装 Java
- https://zhuanlan.zhihu.com/p/137114682
配置路径
# 打开
sudo gedit ~/.bashrc
# 添加自己的路径,格式如下,添加后保存关闭
export PATH=/home/wyl/plink2:$PATH
# 使环境变量生效
source ~/.bashrc
# 关闭此终端,打开新的终端测试
使用PopLDdecay软件绘制LD衰减图
conda install -c bioconda PopLDdecay
PopLDdecay
https://blog.csdn.net/m0_53915752/article/details/137159462
- LD衰减图绘制–PopLDdecay
https://yijiaobani.blog.csdn.net/article/details/127496418
重测序SCI标配——LD衰减图解读
https://zhuanlan.zhihu.com/p/367001288
- LD衰减图的理解与应用
https://www.jianshu.com/p/a36bd4145ef7 - 连锁不平衡以及连锁不平衡衰减
https://www.jianshu.com/p/cd806ebe7d36 - LD: 利用Plink软件进行连锁不平衡计算和绘图
https://blog.csdn.net/qq_40256654/article/details/136394717
数据格式转换
GBS hapmap 格式 转化为Plink格式:tassel vcftools方法汇总
https://wap.sciencenet.cn/home.php?mod=space&do=blog&id=1191913
- 建议使用TASSEl软件读取hapmap格式数据, 另存为Plink格式数据, 这是最简单的
- 使用代码的方法复杂难懂一点


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









