






目录
一、简介
特征提取是指从原始数据中提取出有效的信息,在生信项目中的原始数据一般分为氨基酸序列、核苷酸序列或者蛋白质的三维结构。特征提取的过程指从原始数据中提取出特征并将特征输入到机器学习算法中,以训练出具有预测或分类等功能的模型。
在生信项目中,特征提取过程是非常重要的,这直接影响到后面模型训练的效果,不同的特征提取方法提取到的特征可能会造成最后模型训练结果的不同,也有些方法采取了特征融合的方法,将多维数据特征进行融合以输入模型中。生信项目特征提取的方法是多样化的,本文主要讲解几种常用的传统数据特征提取方法,如K-mer特征提取、CGR特征提取、FCGR特征提取。
二、K-mer特征提取
本章将从两个方面介绍K-mer特征提取,第一方面为介绍K-mer,并介绍如何提取K-mer;第二方面为介绍K-mer频率提取。K-mer特征提取一般是对核苷酸序列的一种特征提取方法,但是氨基酸序列同样适用。本章只针对核苷酸序列做讲解。
①K-mer特征
核苷酸序列是包括A、G、C、T或U(DNA序列是T,RNA序列是U)四种核苷酸的排列组合,若两条序列片段具有相似的组合结构时,则这两个核苷酸片段的相似度程度很高。若下载的核苷酸序列中存在N,则说明该序列碱基N所在的位置没有明确的测序结果。一般情况下,是将这些N碱基随机替换成A、G、C、T或U中的任意一个。
K-mer特征是一种基于窗口的序列特征的表示方法,K-mer特征提取的基本思路是,将长序列分解成固定长度的K-mer单元,即将序列以K个碱基为单位、1个碱基为步长,一次截取获得K-mer。如下图所示为以K=5为例,提取K-mer。
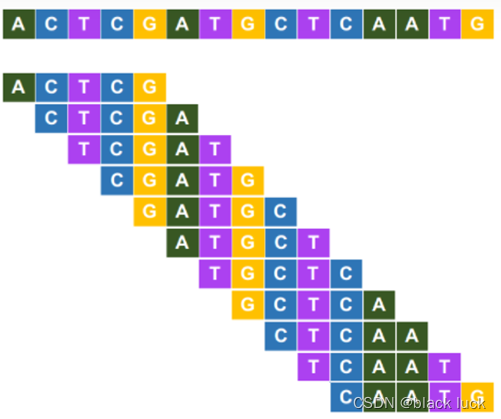
②K-mer频率特征
提取完K-mer特征之后我们需要把K-mer特征转化为计算机可以识别的数值的方式,即K-mer特征向量化。通常我们是将每个K-mer视为一个特征,其出现的频率或计数作为该特征的值,即K-mer频率特征。该特征向量的维度为 维。若共有n条序列,其取K-mer的K为,则该输入特征为(n,256)维的特征。如下为提取4-mer频率特征的Python代码实现方式。
k_list = ["A", "C", "G", "T"]
nucl_list = ["A", "C", "G", "T"]
for i in range(3):
tmp = []
for item in nucl_list:
for nucl in k_list:
tmp.append(nucl+item)
k_list = tmp #输出一个4-mer的K_list
mer2dict = {mer: idx for idx, mer in enumerate(k_list)} #创建一个字典,字典的键是mer,值是这些元素在k_list中的索引idx
file_list = os.listdir(file_in_fn) #获取路径下的所有文件和文件夹的名字,并存储在file_list列表中
num_file = len(file_list) #计算列表中元素的数量
file2idx = {}
feature = np.zeros((num_file, 256)) #创建一个全零矩阵
for idx, file in enumerate(file_list): #遍历文件列表,同时给每个文件一个唯一的索引
file2idx[file.rsplit('.', 1)[0]] = idx #使用file2idx字典存储每个文件的名字和它对应的索引
for record in SeqIO.parse(file_in_fn + file, 'fasta'): #读取Fasta文件
seq = str(record.seq) #转换为字符串
seq = seq.upper() #转换为大写,以确保碱基的一致性
for pos in range(len(seq)-3): #遍历每个序列,以4-mer为窗口
try:
feature[idx][mer2dict[seq[pos:pos+4]]] += 1 #若找到了该索引,则在该位置的计数加1;若pos=0,指从第1个元素开始取到第4个([0,4])
except:
pass #若碱基序列不在字典中,则跳过三、CGR特征提取
CGR(Chaos Game Representation)指混沌博弈表示,这是一种用于表示或分析数据的可视化方法。CGR在生信项目中可以用于描述核苷酸或氨基酸序列,其核心思想是通过一种随机的方式,将一系列的字符串转化为一个平面图像,即将核苷酸序列或氨基酸序列可视化。
CGR最初是因为Sierpinski三角形而提出的,其过程如图所示,随机选择一个起点(S),随机选择一个顶点(A),并在到顶点A距离的一半出绘制点P1。重复此过程,以P1为新的起点。第二个点(P2)在第二个随机选择的顶点(C)的一半绘制,通过重复此过程,最后出现了一个Sierpinski三角形。
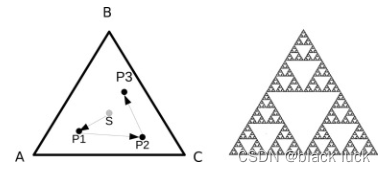
①对核苷酸序列的CGR特征提取
对核苷酸序列的CGR提取并没有选择使用三角形,而是基于正方形,四个顶点带边四个核苷酸,按照上述的方法进行构建出来的CGR。如下图所示,左图为CGR算法流程,右图为由于迭代过程导致的CGR空间划分。
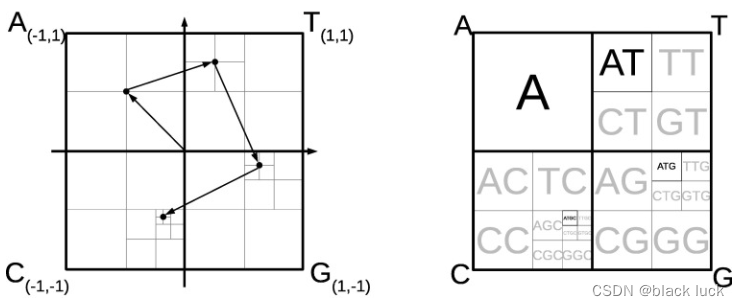
CGR基于几何方法的迭代公式为:
式中,指起点位置,该起点可以随机选择也可以预定义;
指CGR的维度;
指序列S的位置,
指比例因子;当
为T或者G时,
,否则
;当
为A或者T时,
,否则
。
②对氨基酸序列的CGR特征提取
对氨基酸序列的CGR特征提取,即将三角形扩展到更大的多边形中,但对于超过四个顶点,CGR就会变得嘈杂,并且会出现点重叠的现象,为此需要调整比例因子以避免重叠,可以使用如下公式进行调整:
式中,为顶点
的x坐标;
为顶点
的y坐标;
为半径;
为顶点;
为顶点数;
为方向角。
CGR方法的具体代码实现可在CSDN或github上搜索出现,这里不再过多赘述。
四、FCGR特征提取
FCGR(Frequency Chaos Game Representation )指频率矩阵混沌博弈表示,FCGR实现了CGR的抽象表示。FCGR是基于预定义网格计算CGR的带你,如下图所示为K-mer频率K取3时的FCGR图像。左图为CGR,右图为FCGR。

FCGR网格尺寸为;FCGR网络中的象限数为
;其中K为K-mer的K值;Q为象限数。
五、总结
本文主要介绍了几种常用的传统方法的特征提取流程,但特征的选择应保持具体项目具体分析的原则,对于机器学习的特征提取流程会在以后得文章中进行展示。

















 512
512

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









