






一.R环境初识
1.1R环境结合了:
- 开源,可扩展的环境
- 简单有效的编程语言
- 数据处理,清理和存储
- 统计与可视化
1.2R语言的集成开发环境(狭义RStudio)
- 图形界面
- 优秀的学习工具
- 科研用途免费
- 开源
1.3如何在RStudio中新建项目 --R project-->R Script
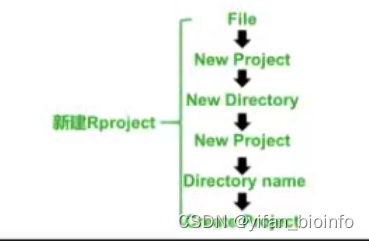
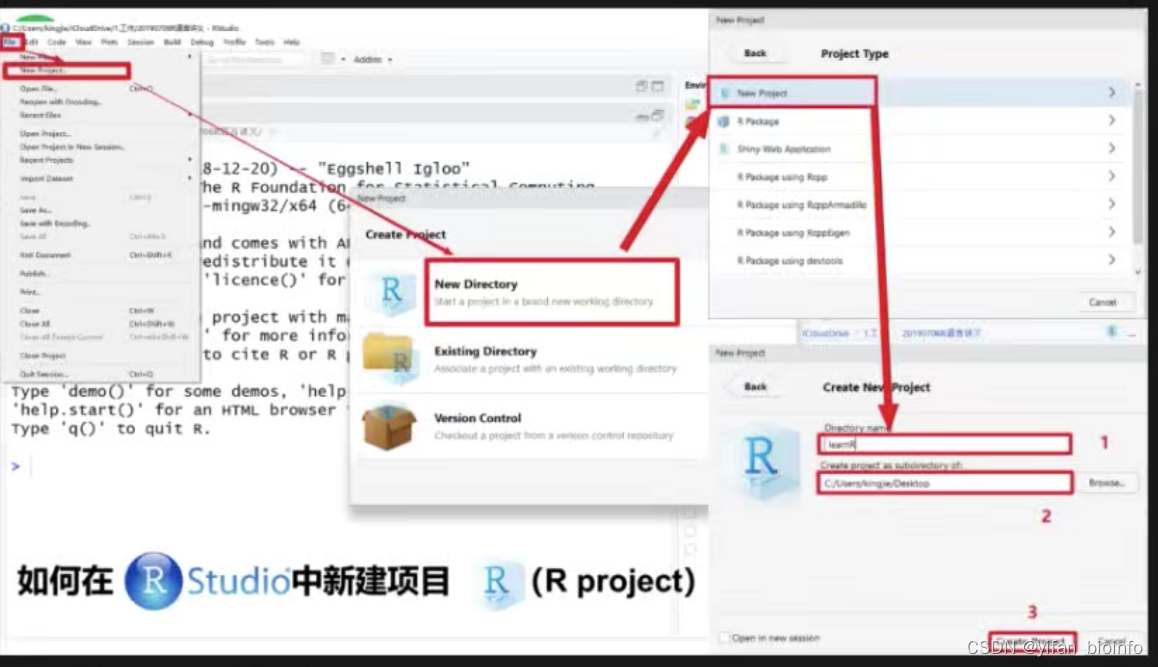
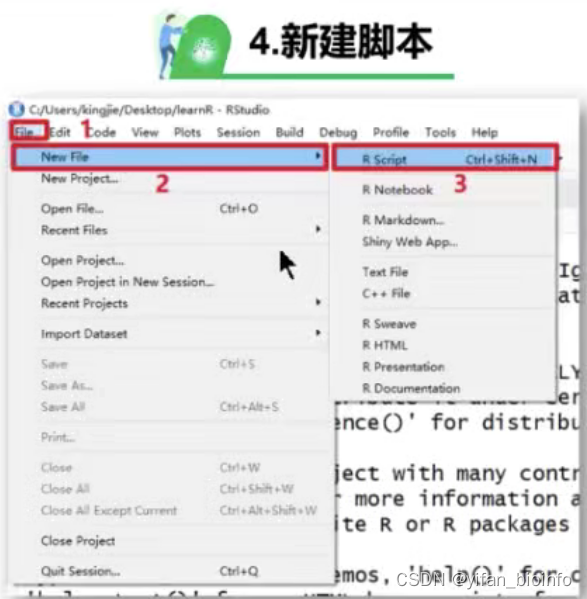
tip:如果你足够理解R的项目创建以及管理逻辑,你就不会选择直接打开一个RScript,这会导致你的工作目录在一个任意的位置,会增加你对R文件的管理难度。每一个RScript都有自己归属的RProject,你应该先打开它,再选择你需要使用的RScript。
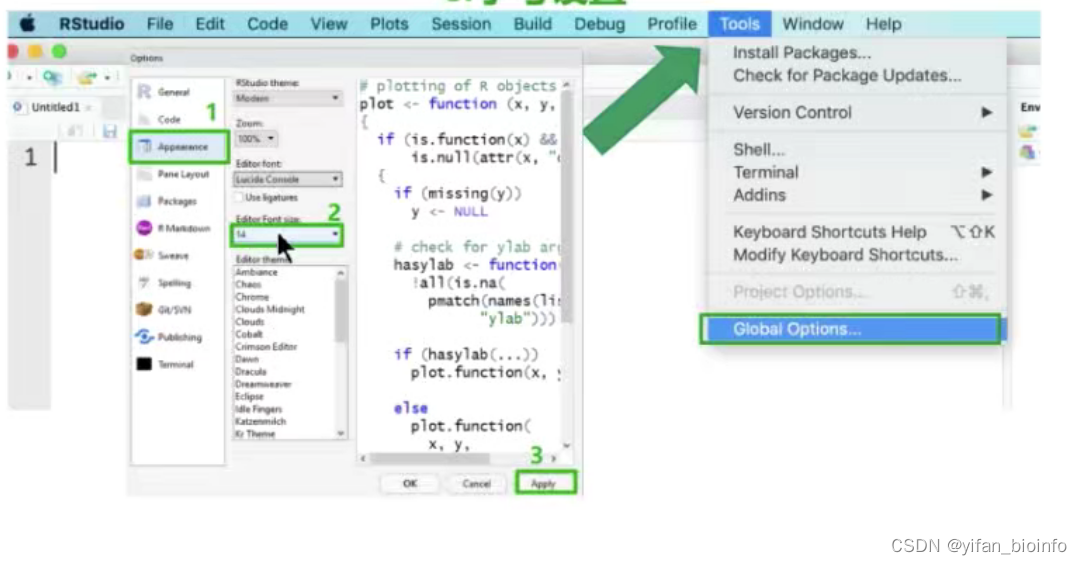
1.4R Studio界面介绍
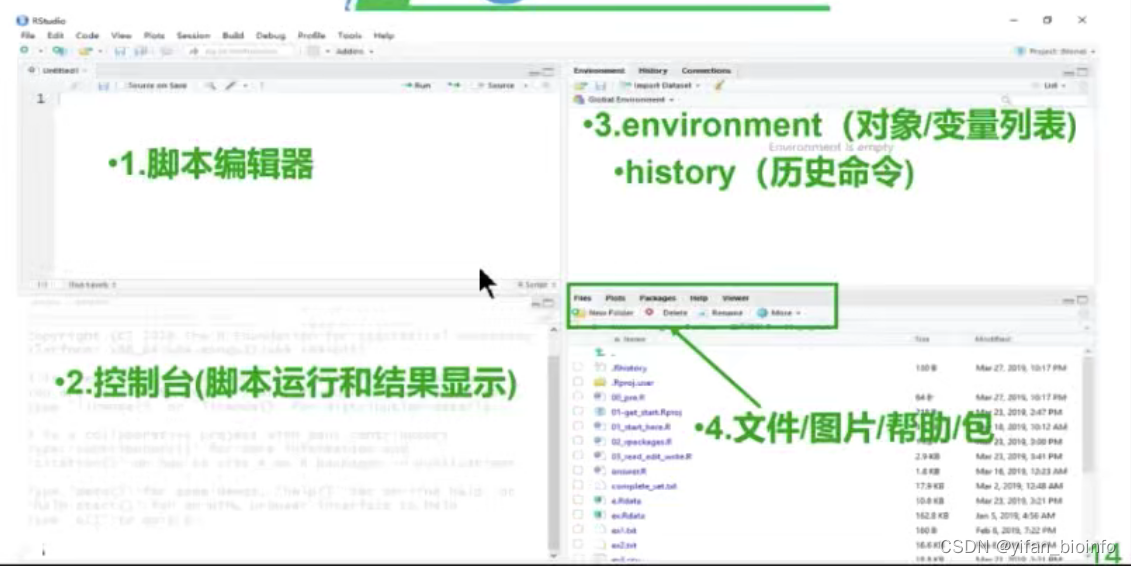
1.4.1界面设置-->字号设置...
1.5与R“交互”
1.5.1交互式会话(session)--了解session
用户-->发送命令
R-->执行并返回结果
举个简单的例子说明session:我们常用的word软件可以同时打开多个窗口(文件),就像RStudio可以同时打开多个session一样。用户对每个独立的session进行设置,session之间互不干扰。由下图可以看出,用户可以对session进行诸如,重启退出等多种操作。

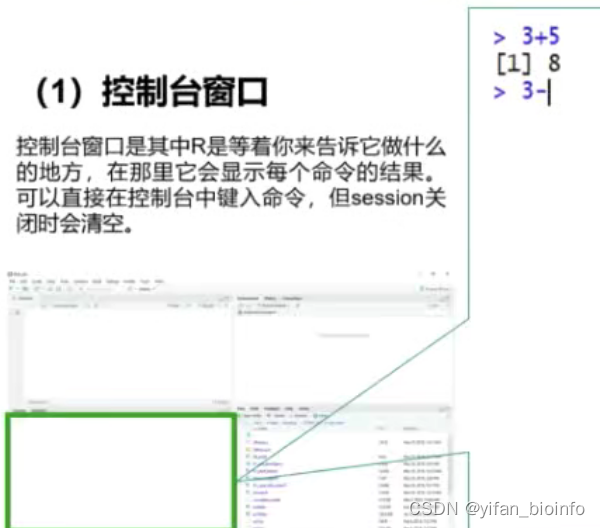
1.5.2 与R“交互”的两种方式
- 控制台窗口:仅限一次命令
- 脚本编辑器
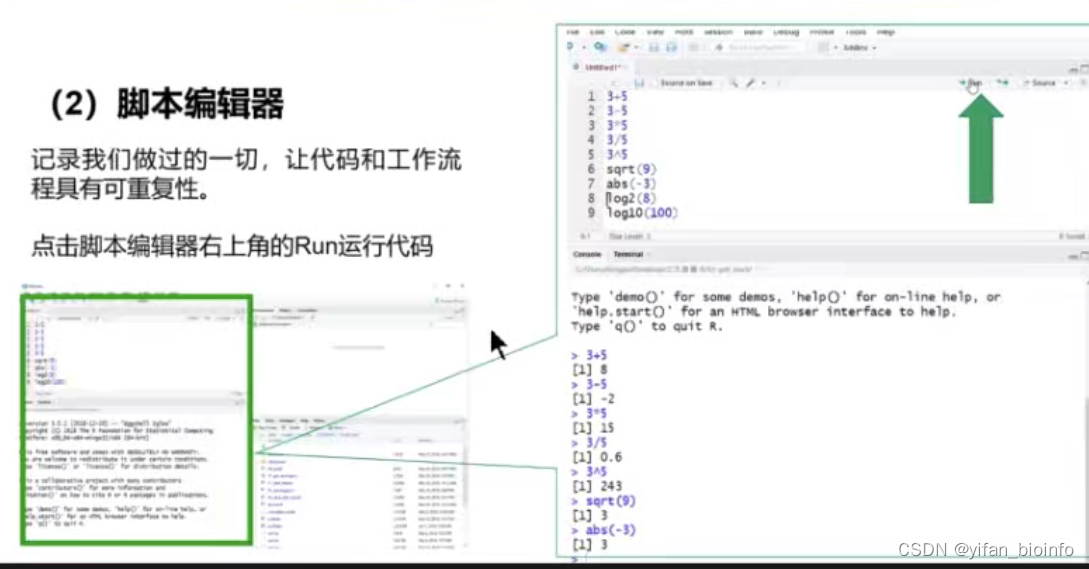
1.5.3脚本的运行方式
- 逐行运行
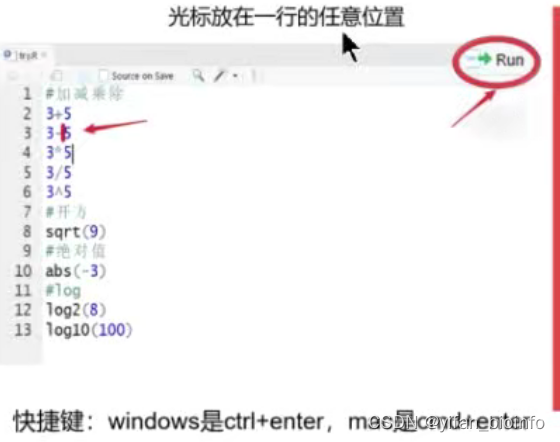
- 选中运行
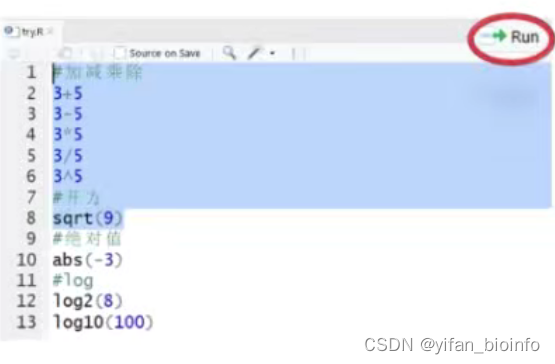
1.5.4脚本的注释
-
使用“#”为脚本添加注释 。让自己和其他协作者了解代码的用途。
-
R自动忽略运行“#”右侧的任何内容。
1.5.5 脚本的保存和关闭(工作目录)
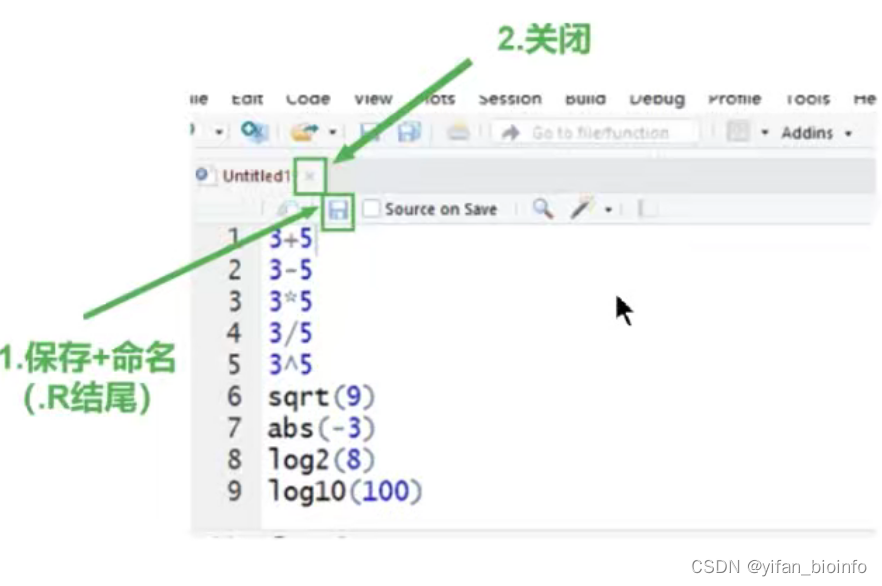
- 工作目录
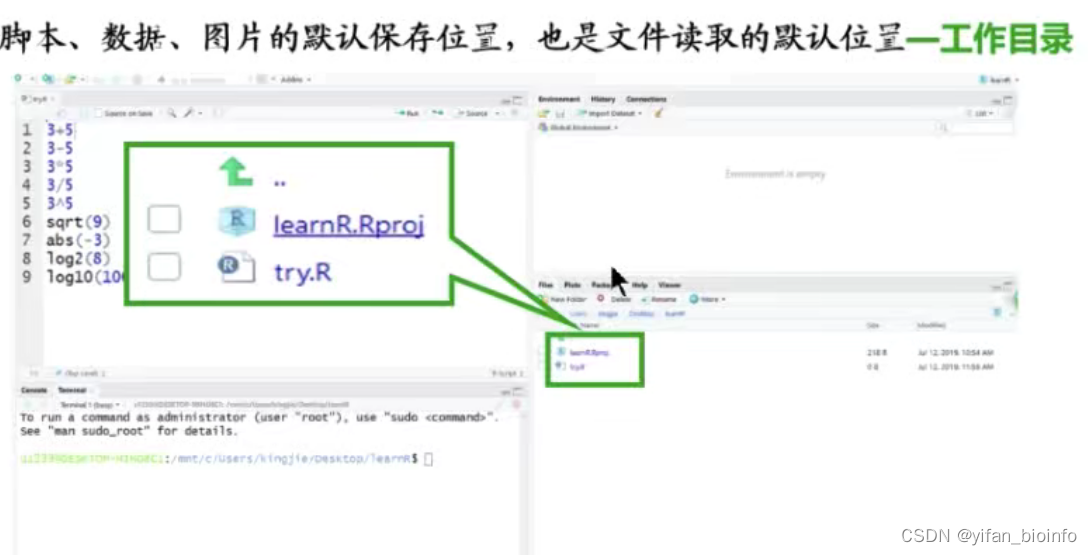
1.5.6R语言的几类命令响应
- 正确示例
- 错误情况

- 警告(脚本输出结果非错即对,忽略warning)
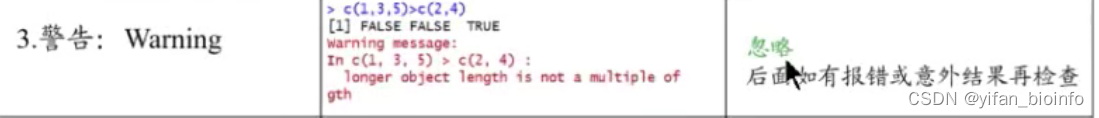
- ''没反应''
当控制台出现“>”提示,表示R等待用户输入下一条命令,此时我们默认上一条命令已经完成

- 命令正在运行
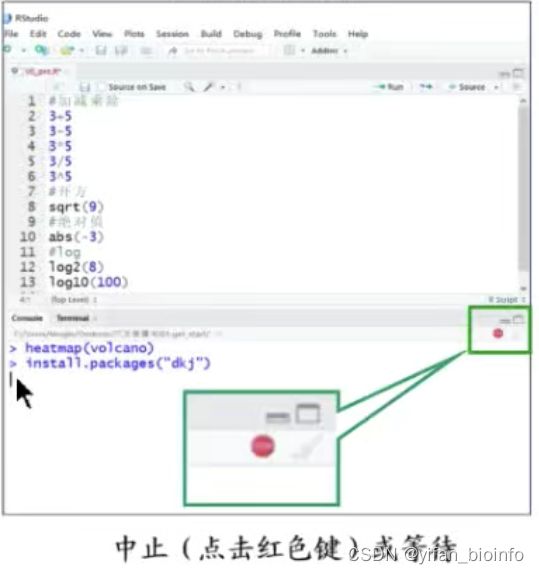
如果一条命令运行了很久还没有完成,可以选择点击“stop”终止,卡死的情况可以重启当前session
- 命令不完整-->出现“+”
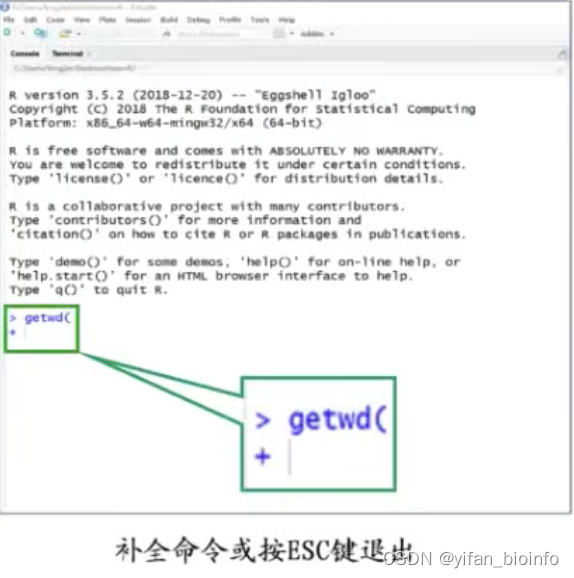
- 出现提示信息
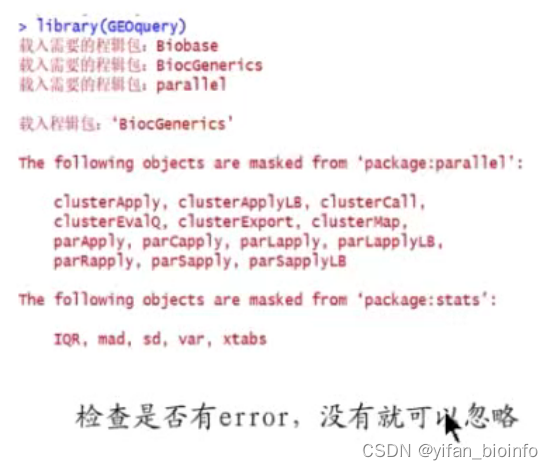
- 进一步选择

在用户安装R包的时候多半会被R提问,是否更新R包?建议输入n,然后回车。
二 .数据类型和向量
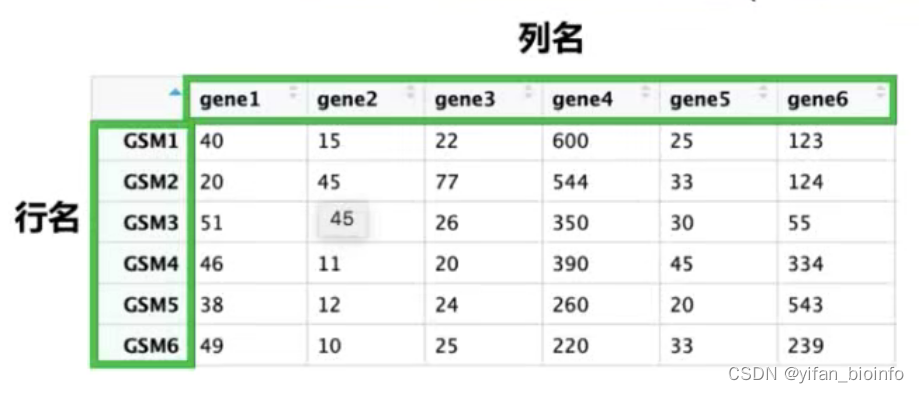
2.1从表格看数据类型
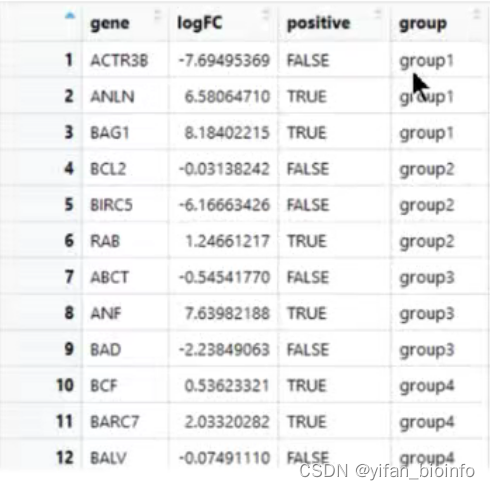
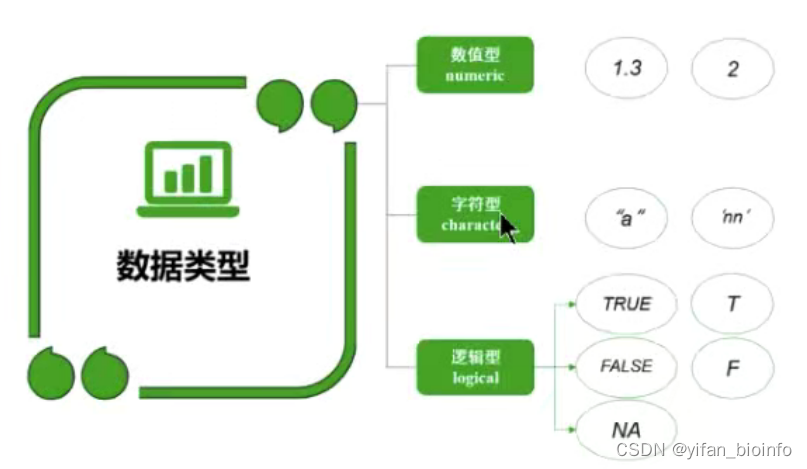
- 数值型(numeric)字符型(character)逻辑型(logical)
判断数据类型的函数:class(),将要判断的内容写在括号里

- 常见报错
tip:多用快捷键,程序员要学会“偷懒”-->Tab''补全键"

tip: 善用键盘
2.2.1逻辑性数据

2.2.2数据类型的判断和转换

2.2.3多个数据如何组织

-
打开已有的Rproject
- 切换Rproject
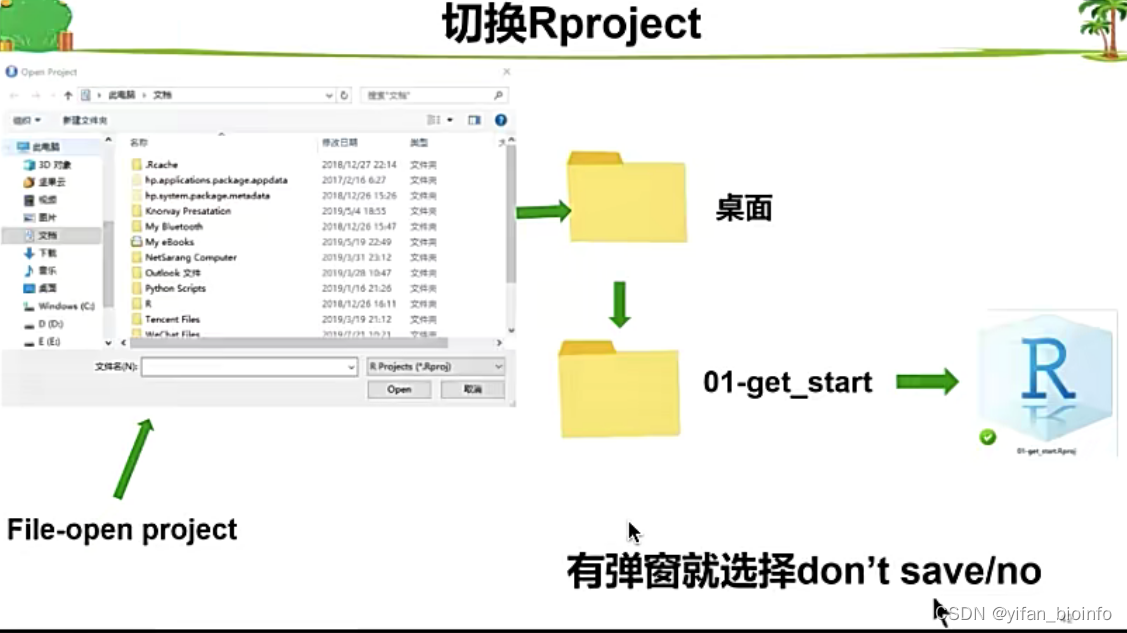
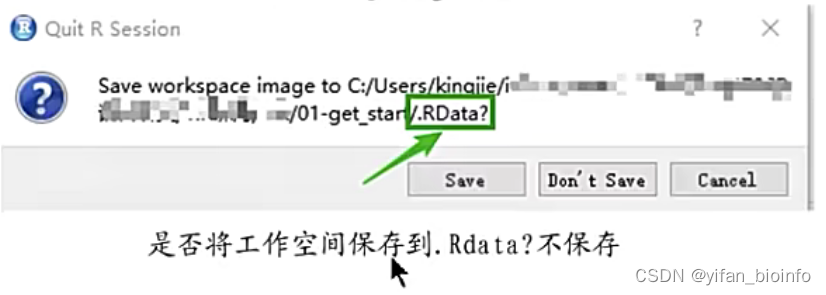
tip:为了节省空间,不要保存缓存文件
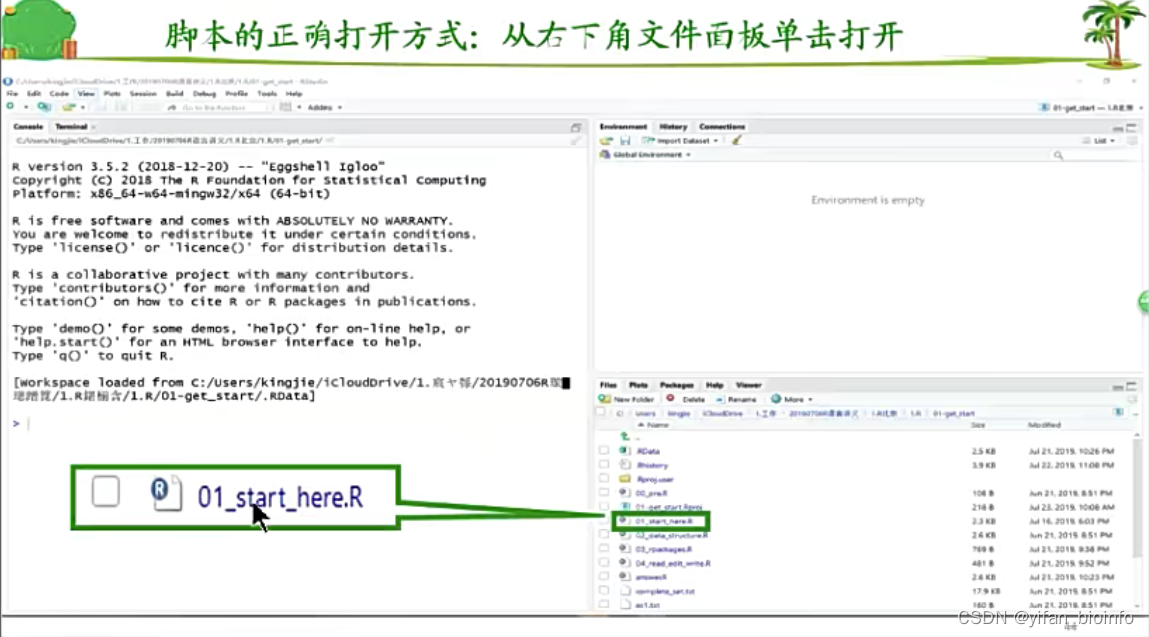
- 脚本是乱码的解决方案
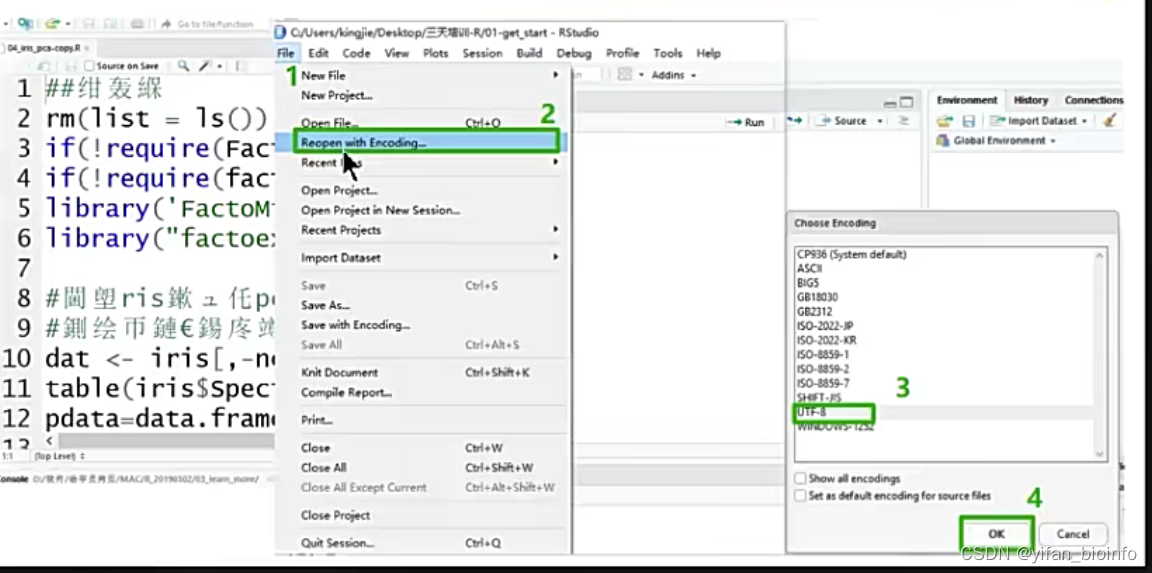
出现乱码通常是因为编码方式不同
2.2向量的生成

tip:数据类型转换的优先顺序-->向量只允许有一种数据类型
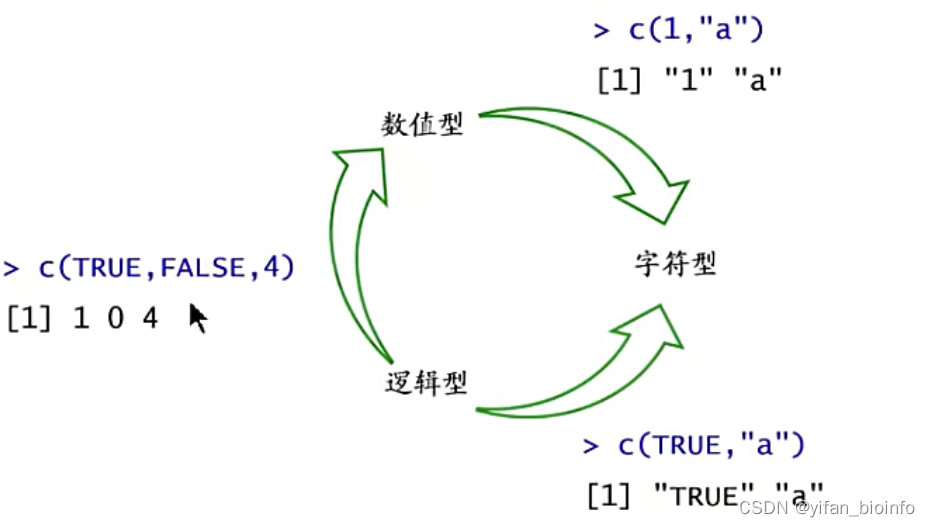

2.3对单个向量进行的操作
- 赋值

术语: 向量的 下一级是元素(x向量的元素有1,3,5)
- 简单数学计算
tip:R语言是直接可以对向量进行操作的,所以与其费劲心思的想怎么写循环语句,倒不如对向量(包含多个元素)进行操作。举例:下图中x向量有四个元素,所以直接对向量x执行各种操作即可,不用写循环处理四遍。
术语:向量中元素的个数称为向量的长度

- 根据某条件进行判断,生成逻辑值向量

- 初级统计函数

2.4对两个向量进行操作



- x和y不一样长:理解一下循环补齐 (无前后顺序,短的向量补齐长的向量)
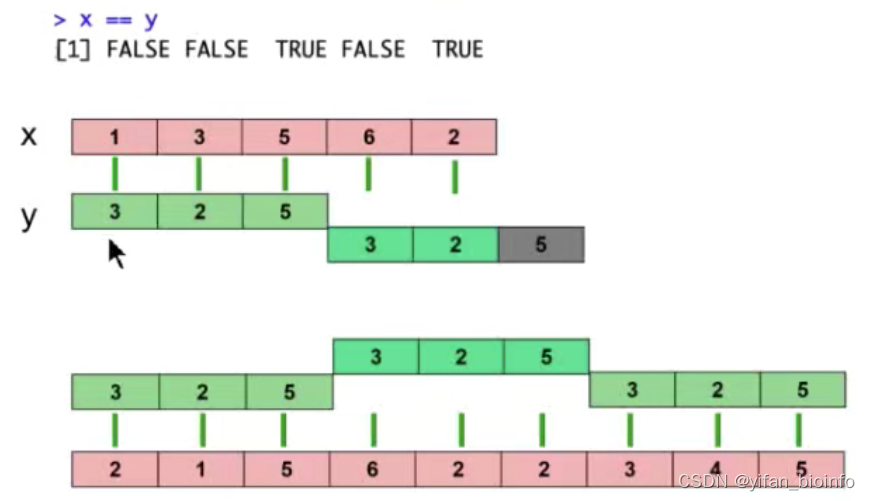
- 利用循环补齐简化代码

2.5向量筛选(取子集)
- []:将TRUE对应的值挑选出来,FALSE丢弃
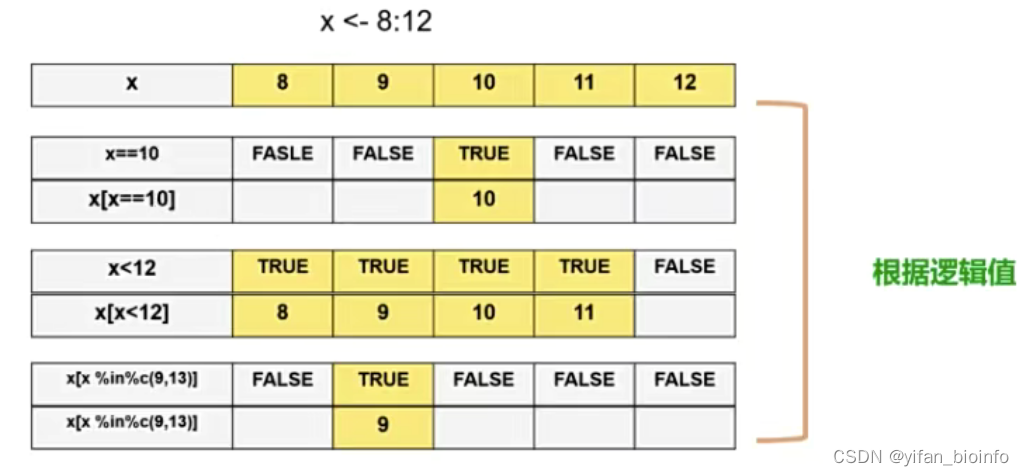
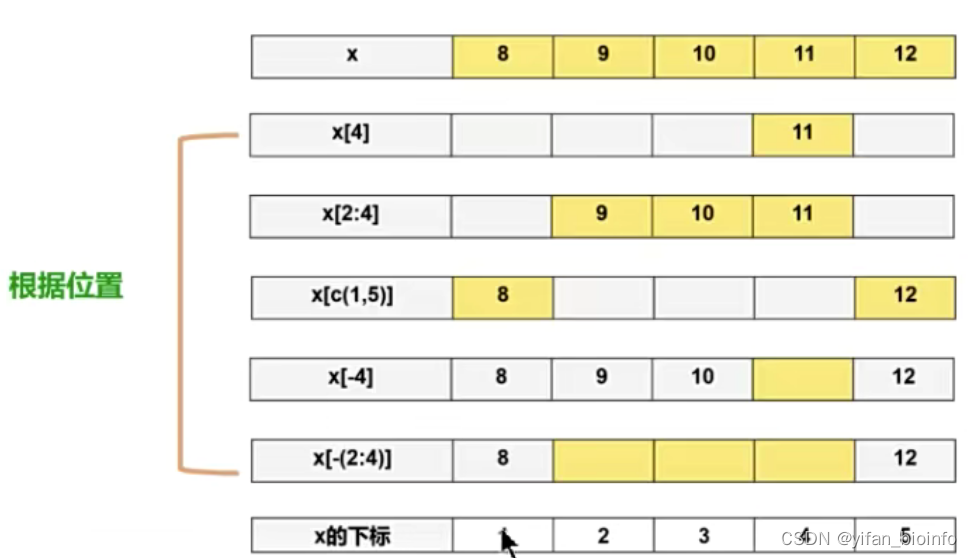
2.6如何修改向量中的某个/某些元素
- 取子集+赋值
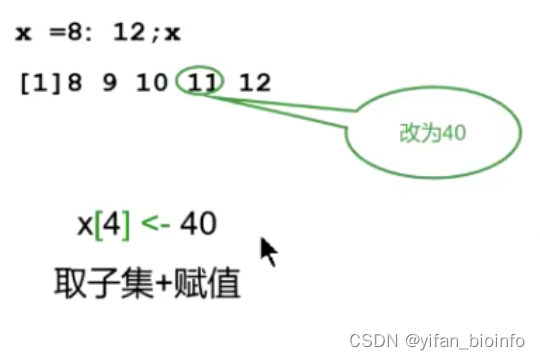
- 按照逻辑值

#练习
# 1.将基因名"ACTR3B","ANLN","BAG1","BCL2","BIRC5","RAB","ABCT","ANF","BAD","BCF","BARC7","BALV"组成一个向量,赋值给x
x <- c("ACTR3B","ANLN","BAG1","BCL2","BIRC5","RAB","ABCT","ANF","BAD","BCF","BARC7","BALV")
# 2.用函数计算向量长度
length(x)
# 3.用向量取子集的方法,选出第1,3,5,7,9,11个基因名。
x[c(1,3,5,7,9,11)]
# 4.用向量取子集的方法,选出除倒数第2个以外所有的基因名。
x[-(11:12)]
# 5.用向量取子集的方法,选出出在c("ANLN", "BCL2","TP53")中有的基因名。
# 提示:%in%
x[x %in% c("ANLN", "BCL2","TP53")]
# 6.修改第6个基因名为"a"并查看是否成功
x[6] <- "a"
x
#7.生成100个随机数: rnorm(n=100,mean=0,sd=18)
y <- rnorm(n=100,mean=0,sd=18)
#将小于-2的统一改为-2,将大于2的统一改为2
y[ y < -2 ] <- -2
y[ y > 2 ] <- 2
y2.7数据类型--矩阵
- 从一个向量得到矩阵
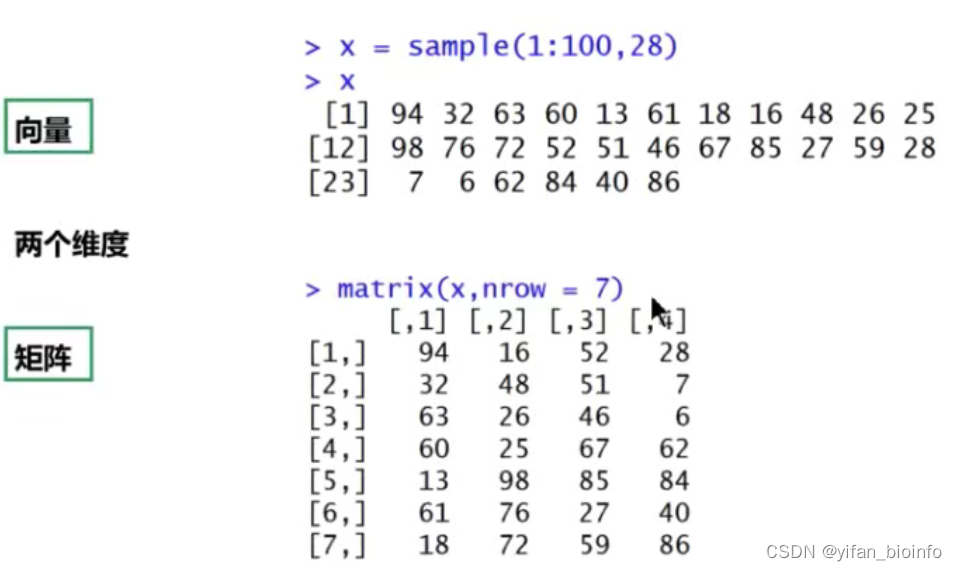
- 从多个向量合并得到矩阵

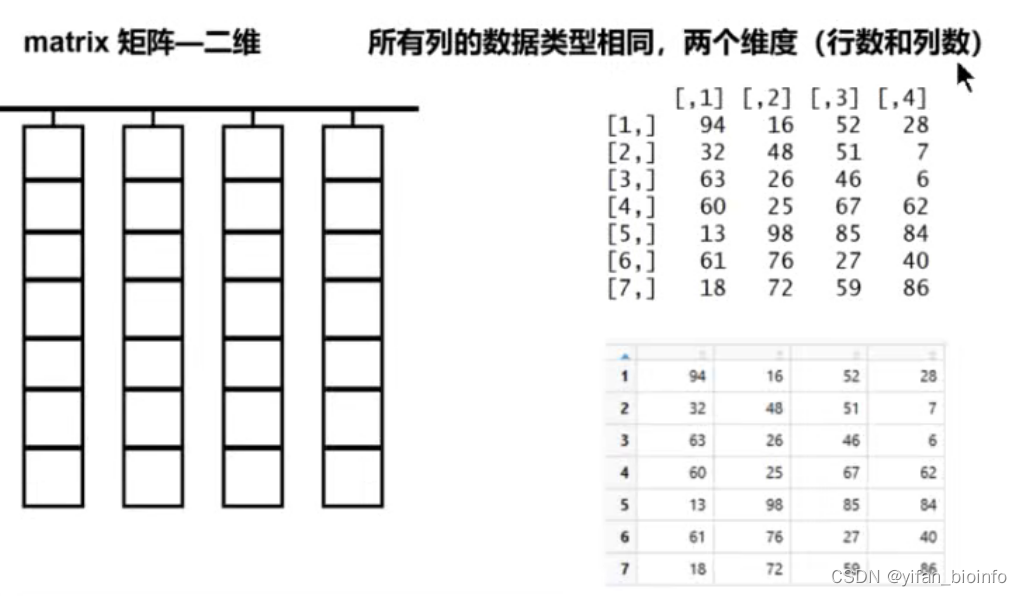
tip:向量的维度是一维用长度衡量(向量中元素的个数即就是长度);矩阵是二维的,两个维度(行数和列数)
下图展示在RStudio中两种不同的矩阵展示形式
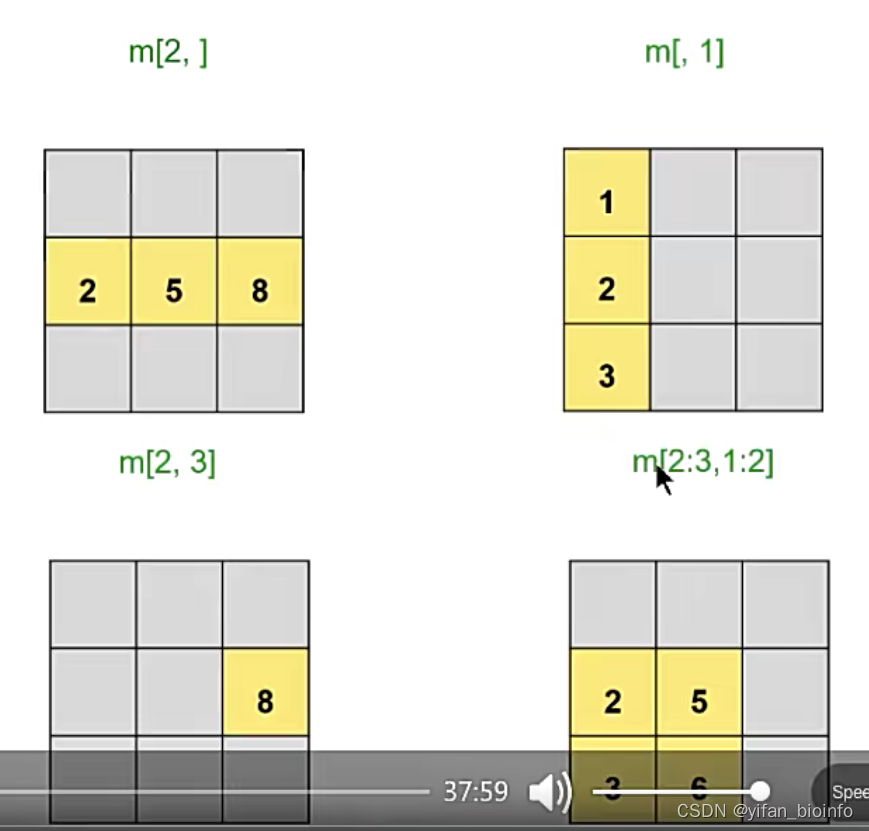
2.7.1矩阵新建和取子集

2.7.2矩阵的转置和转换

2.7.3矩阵画热图

热图:颜色的深浅表示数值的大小
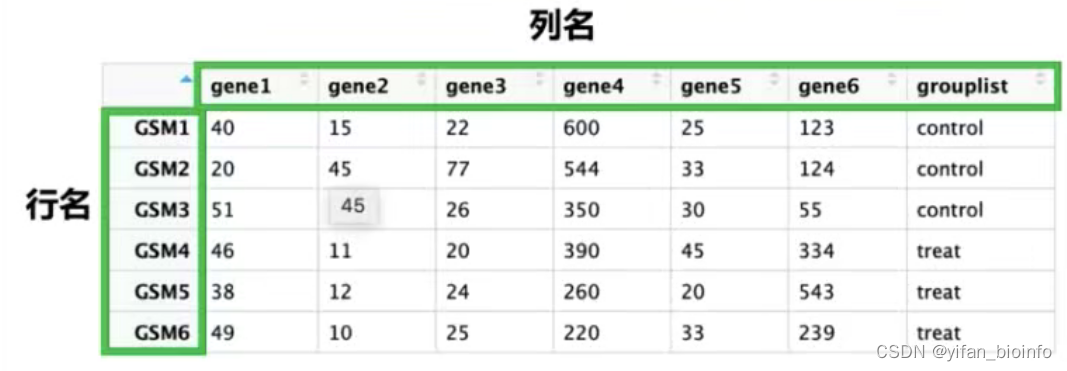
2.8数据类型--数据框
重点学习数据框,作图处理的数据绝大多数都是数据框

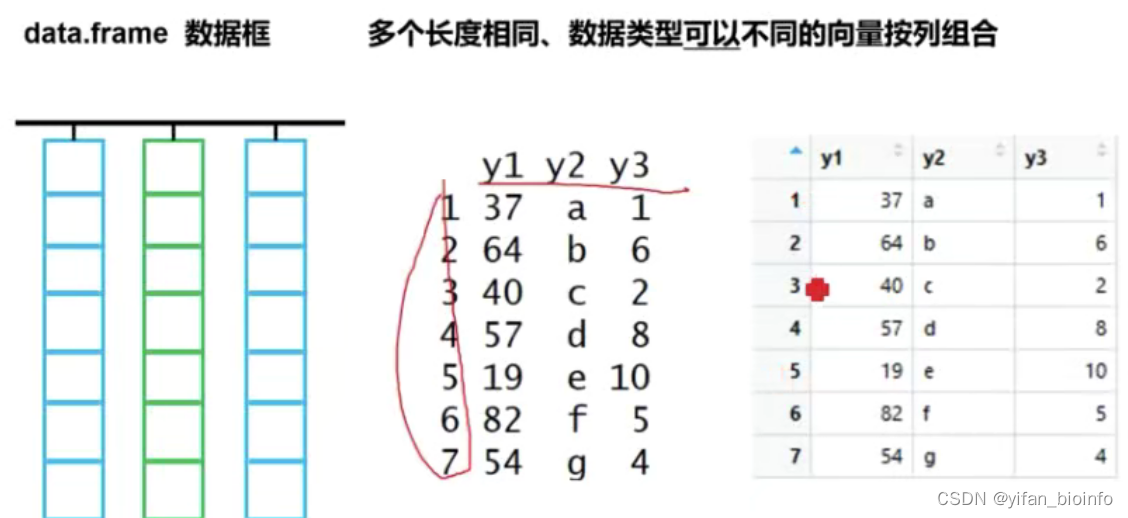
2.8.1数据框来源

2.8.2数据框取子集
- 取一个

- 取一行或者一列,行列双选
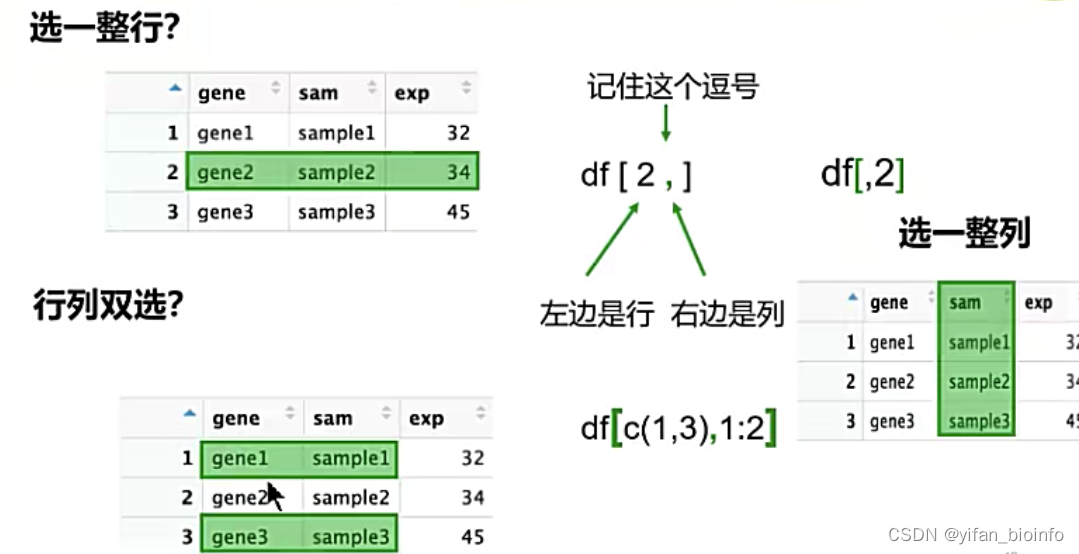
tip:每一个维度如果牵扯多行和多列,必须要将这个维度表示为向量。
- 根据行名或者列名提取

- 进阶--理解以下代码
将嵌套的代码写成上下结构会更好一些(第一二行为上下结构,第三行是嵌套结构)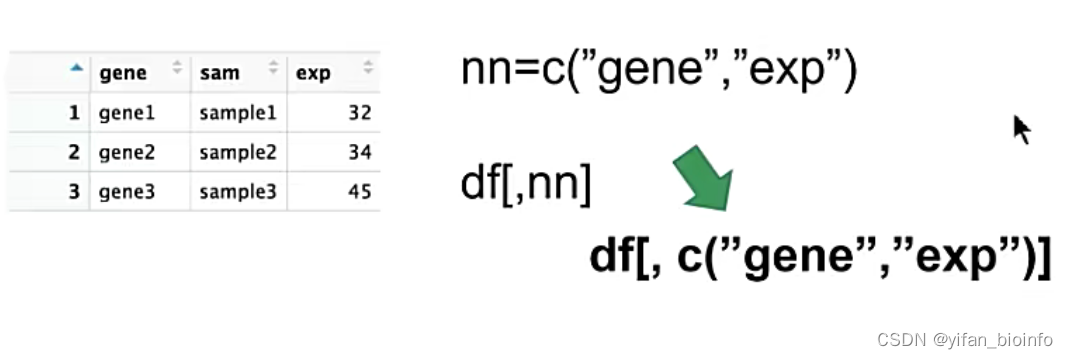
要习惯以函数返回值取数据,不要主观臆断
以下代码表示取数据框第三列,-表示反选,不要第三列
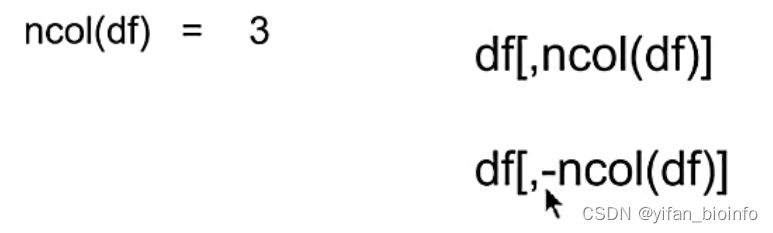
2.8.3提取列的常用操作
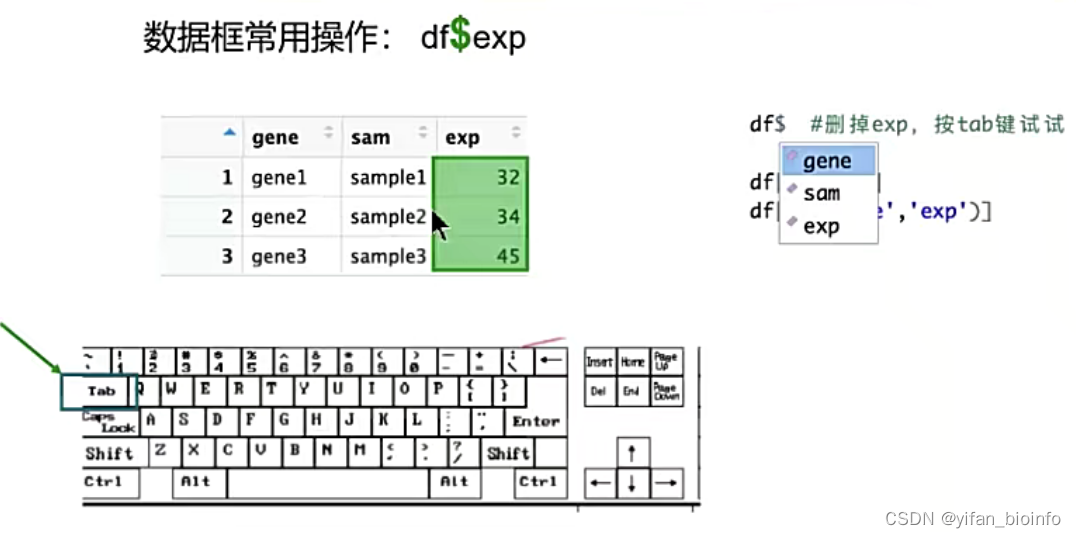

2.8.4数据框的编辑
- 改内容

不存在列名为abc那列,上述代码执行完之后会新增这一列
- 改行名和列明

2.8.5误操作
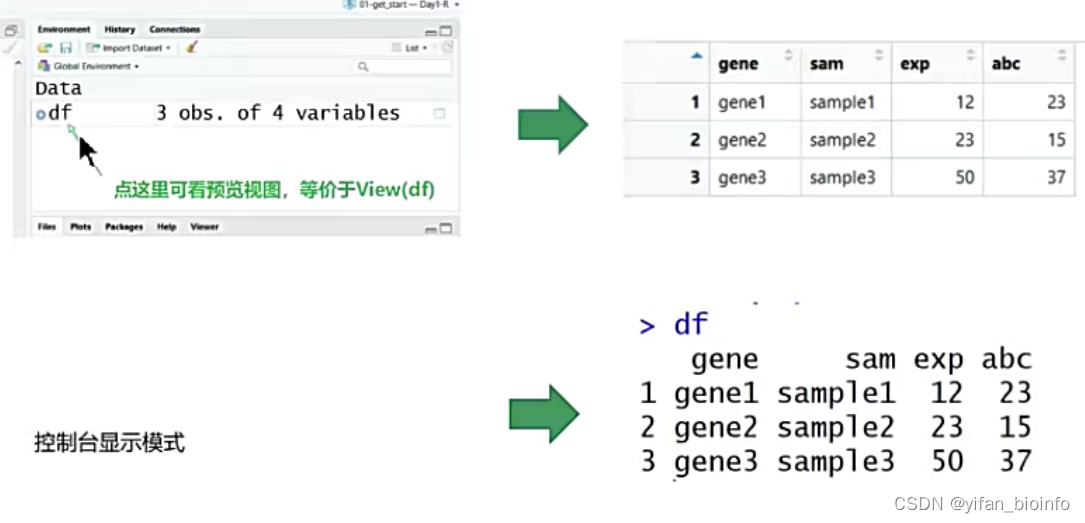
- 数据框的两种形式
2.8.6数据框进阶



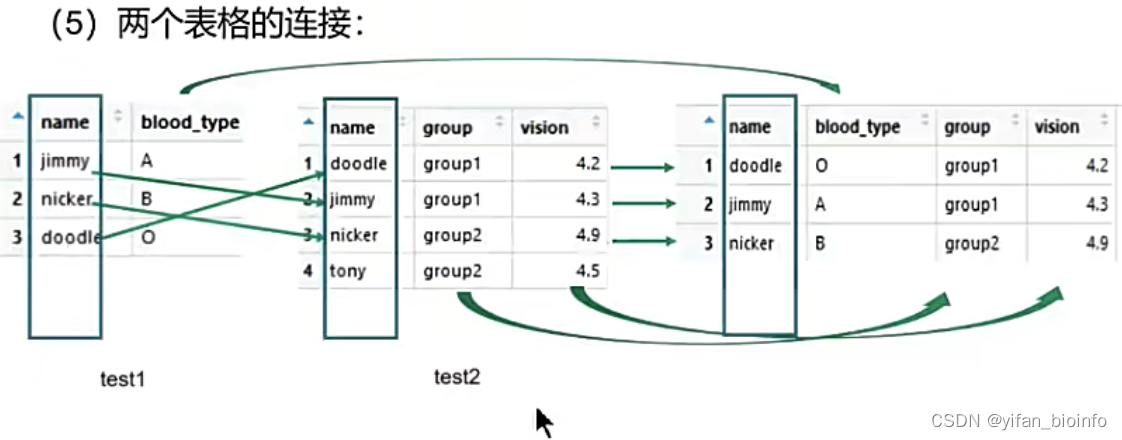
merge(test1,test2,by="name"),name是共同的列名

2.8.7示例代码
#重点:数据框
#1.数据框来源
# (1)在R中新建
# (2)由已有数据转换或处理得到
# (3)从文件中读取
# (4)内置数据集
#2.新建和读取数据框
#options()为全局设置每次在新建数据框时都要先写上这句不然就会牵扯因子很麻烦
options(stringsAsFactors = FALSE)
df <- data.frame(gene = c("gene1","gene2","gene3"),
sam = c("sample1","sample2","sample3"),
exp = c(32,34,45))
df
df <- data.frame(gene = paste0("gene",1:3),
sam = paste0("sample",1:3),
exp = c(32,34,45))
df
df2 <- read.csv("gene.csv")
df2
#3.数据框属性描述
#
dim(df)
nrow(df)
ncol(df)
#
rownames(df)
colnames(df)
# 练习
# 1.新建图示数据框,赋值给test1
# (提示:后三列是用函数生成的随机数,
# 数值不必完全和我截图的相同)
test1 <- data.frame(gene = paste0("gene",1:15),
s1 = rnorm(15),
s2 = rnorm(15),
s3 = rnorm(15))
test1
# 2.读取excise.csv这个文件,赋值给test2。
test2 <- read.csv("excise.csv")
test2
# 3.使用函数,查看test2的列名。
colnames(test2)
# 4.使用函数,查看test2的行数和列数。
ncol(test2)
nrow(test2)
dim(test2)
#4.数据框取子集
df[2,2]
df[2,]
df[,2]
df[c(1,3),1:2]
df[,"gene"]
df[,c('gene','exp')]
df$exp #删掉exp,按tab键试试
mean(df$exp)
#5.数据框编辑
#改一个格
df[3,3]<- 5
#改一整列
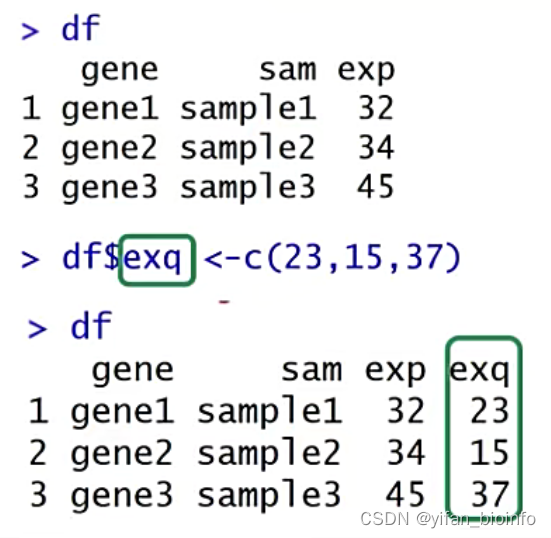
df$exp<-c(12,23,50)
#?
df$abc <-c(23,15,37)
df
#改行名和列名
rownames(df) <- c("r1","r2","r3")
#只修改某一行/列的名
rownames(df)[2]="x"
# 练习
# 1. test = read.csv(“excise.csv”),提取test的第二行
test = read.csv("excise.csv")
test[2,]
# 2.提取第3行第4列
test[4,3]
# 3.求第二列数值的中位数
median(test$s1)
# 4.按照列名,同时提取s1,s3两列。
test[,c("s1","s3")]
# 5.修改后三列的列名为“sample1”,“sample2”,“sample3”
colnames(test)[(ncol(test)-2):ncol(test)] <- paste0('sample',1:3)
# 6.提取sample3列数值大于0的所有行
test[test$sample3 > 0,]
#6.数据框进阶
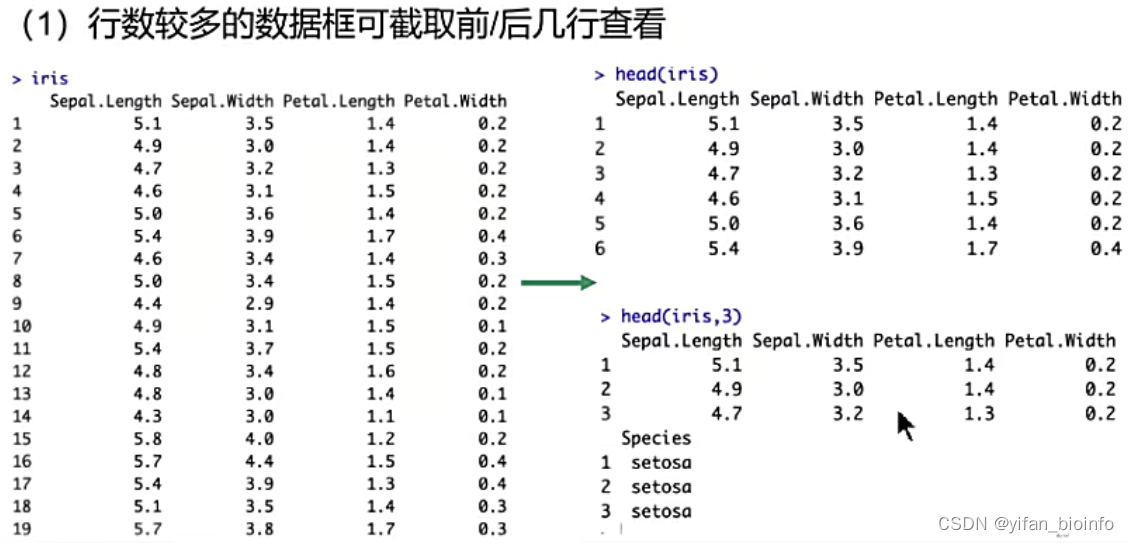
#(1)行数较多的数据框可截取前/后几行查看
iris
head(iris)
head(iris,3)
tail(iris)
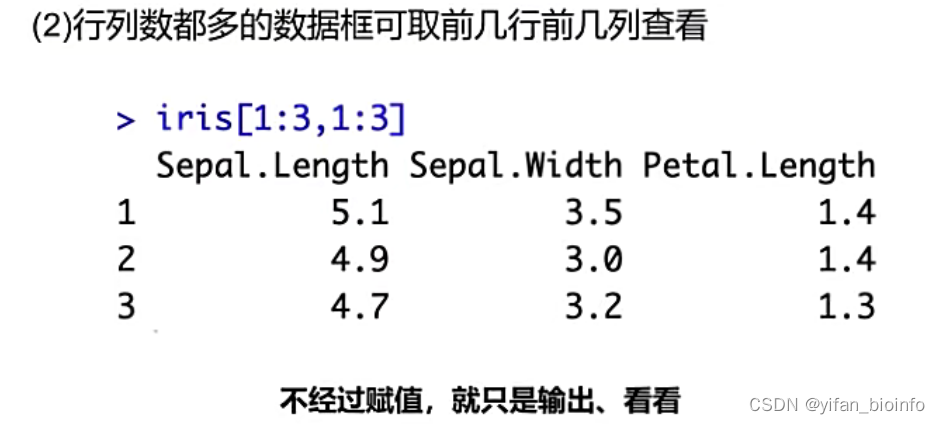
#(2)行列数都多的数据框可取前几行前几列查看
iris[1:3,1:3]
#(3) 查看每一列的数据类型和具体内容
str(df)
str(iris)
#(4)如果列名顺序错乱,如何按照指定顺序重排?
#https://www.jianshu.com/p/09a22a1cdcd1
#(5)去除含有缺失值的行
#生成一个有NA的数据框
df<-data.frame(X1 = LETTERS[1:5],X2 = 1:5)
df[2,2] <- NA
df[4,1] <- NA
df
na.omit(df)
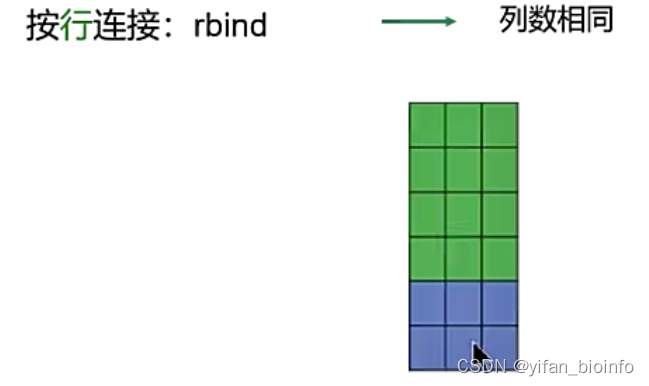
#(6)两个表格的链接
test1 <- data.frame(name = c('jimmy','nicker','doodle'),
blood_type = c("A","B","O"))
test1
test2 <- data.frame(name = c('doodle','jimmy','nicker','tony'),
group = c("group1","group1","group2","group2"),
vision = c(4.2,4.3,4.9,4.5))
test2
test3 <- data.frame(NAME = c('doodle','jimmy','lucy','nicker'),
weight = c(140,145,110,138))
merge(test1,test2,by="name")
merge(test1,test3,by.x = "name",by.y = "NAME")
#####矩阵和列表
m <- matrix(1:9, nrow = 3)
colnames(m) <- c("a","b","c") #列名
m
#整行
m[2,]
#整列
m[,1]
#单个格
m[2,3]
#多个格
m[2:3,1:2]
#转置和转换
m
t(m)
as.data.frame(m)
#列表
l <- list(m=matrix(1:9, nrow = 3),
df=data.frame(gene = paste0("gene",1:3),
sam = paste0("sample",1:3),
exp = c(32,34,45)),
x=c(1,3,5))
l
l[[2]]
l$df
#补充:元素的名字
#(1)向量
x=1:10
names(x)=letters[1:10]
x
x["a"]
#(2)数据框
df
names(df)
df[,"X1"]
#(3)列表
names(l)
l[["df"]]
#删除
#删除一个
rm(l)
#删除多个
rm(df,m)
#删除全部
rm(list = ls())
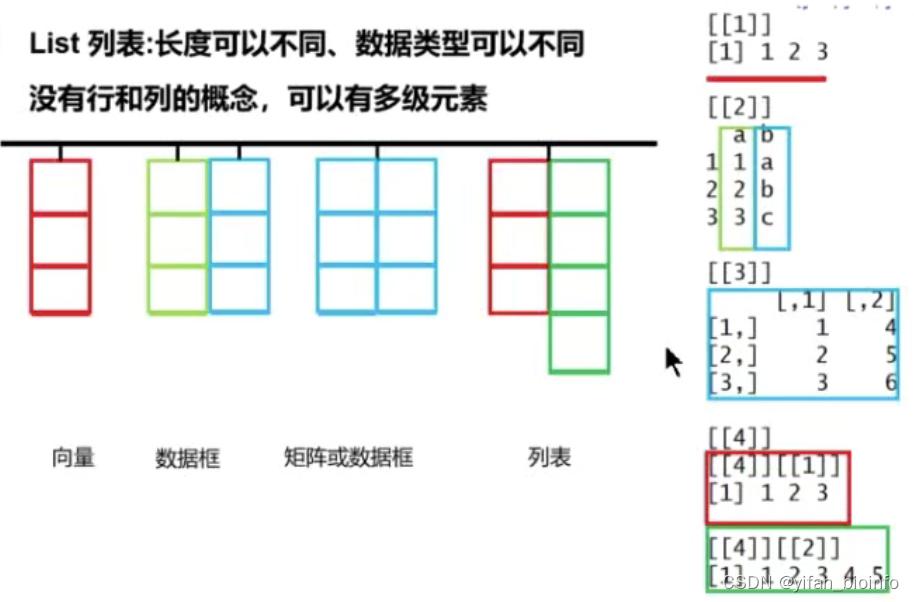
2.9数据类型--列表
exercise:判断数据结构?
矩阵或数据框
矩阵和数据框都有可能
需要赋值,然后用class()判断
真相是

2.9.1列表新建和取子集
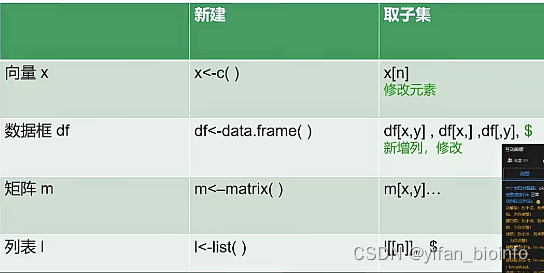
- 新建:列表的下一级是元素:新建的这个列表有三个元素m,df,x


- 取子集
l[[2]]:按照位置取 按照名字取:l$df
- 删除变量
2.10数据结构的总结
- 补充

- 数据结构练习题
#1. 统计iris最后一列有哪几个重复值,分别重复了多少次
options(stringsAsFactors = FALSE)
lastline <- iris[[ncol(iris)]];unique(lastline);table(lastline)
#2.提取iris的前4列,并转换为矩阵,赋值给test。
class(iris[,1:4])
test <- as.matrix(iris[,1:4])
#3.将test的行名改为flower1, flower2...flower149,flower150。
rownames.test <- paste0("flower",1:150)
#4.将test的第51到100行删除
test[-(51:100),]
#5.将iris和test组成一个列表,赋值给tl
tl <- list(iris,test)
#6.提取tl的第二个元素
tl[[2]]
#7.修改tl第二个元素的名字为td
names(tl)[2] <- "td"
三.函数和R包
3.1R创建html文件

注:md就是markdown格式
Rmarkdown为跨平台展示R代码和结果提供了很好的工具。
3.2函数
3.2.1函数与参数
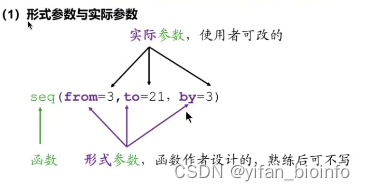

注:写函数的函数function,函数名g
- 绘图函数plot()
思路:将向量x用plot函数生成图
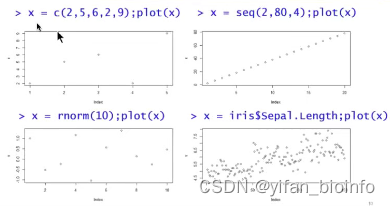
思考:在上述坐标系中,显然有横纵两个向量,只采用一个向量,为什么也可以作图呢?
这个向量表示在纵轴上,横轴是元素的位置,用向量可以表示为c(1:length(x))
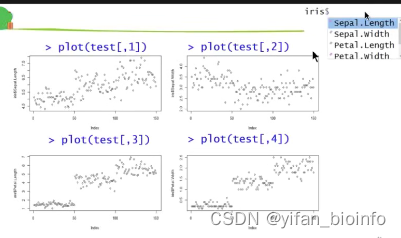
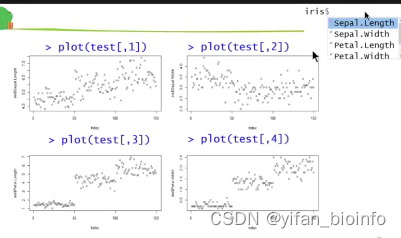
重复写了四遍的代码,不考虑写成函数或者循环吗?
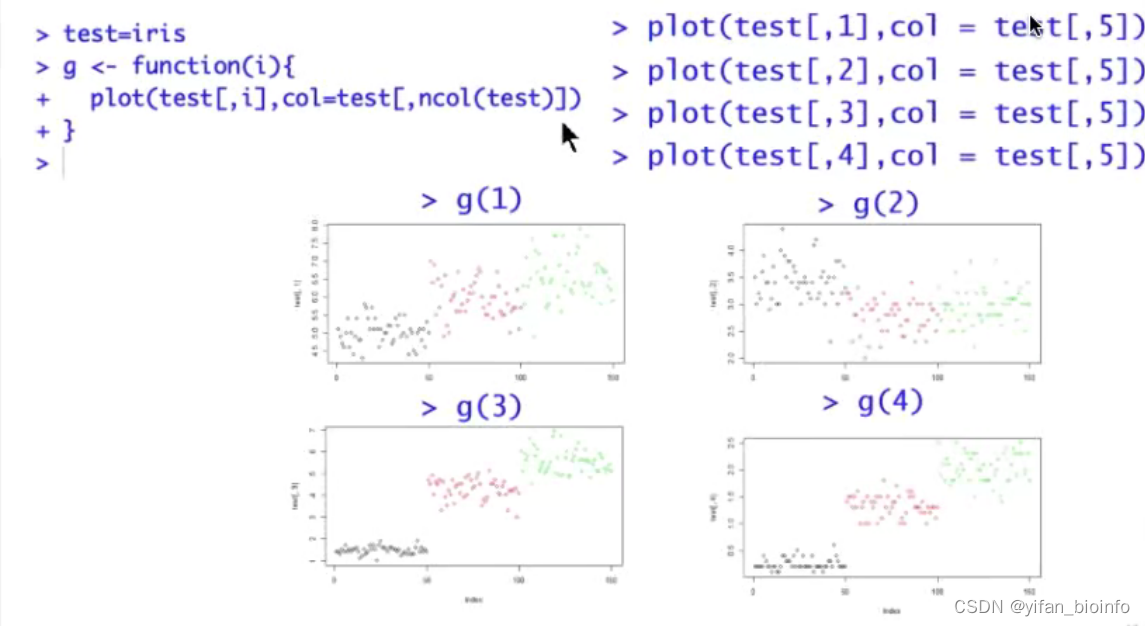
- 默认参数

- 练习

#1.ACE
lenm <- function(x){
c( median(x),mean(x))
}
lenm(iris$Sepal.Length)3.3R包
3.3.1R包介绍


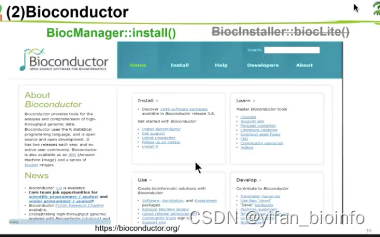
灰色部分的R包安装方法已经过时了,如果在作者的R包中看到请替换为绿色部分
- summary

注意:R包只安装一次,但每次都需要加载
- 解决下载网络问题

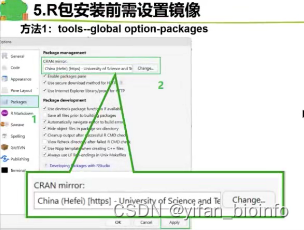
3.3.2R包安装和使用的逻辑


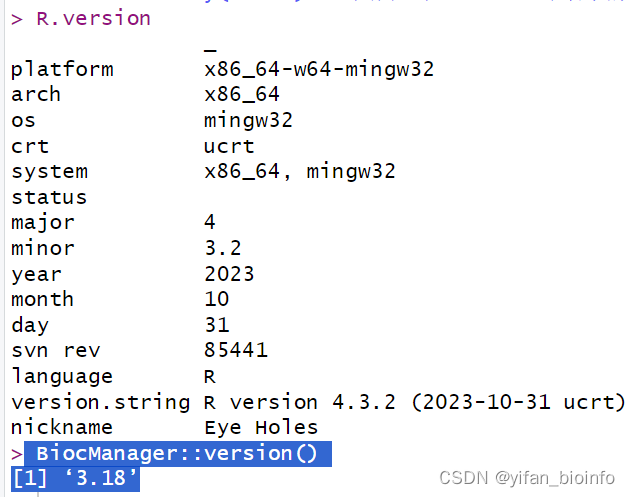
- 包安装失败-->版本不兼容问题
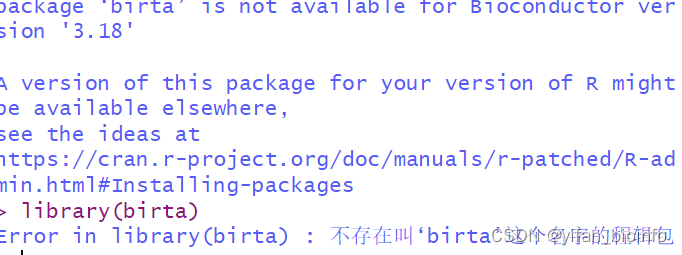 查看版本:
查看版本:
通过goole,csdn,Bioconductor等寻找匹配版本,由于这个包我目前还用不到就不在此花费精力了。
这是CRAN网站的安装版本说明可供参考

- 常见疑问
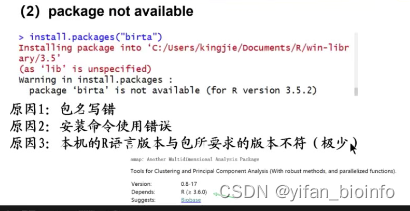

懒得回答:update=F ask=F
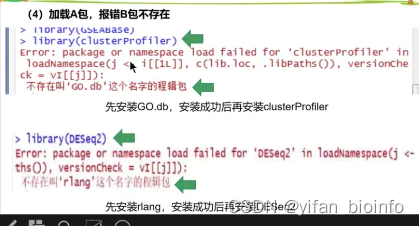


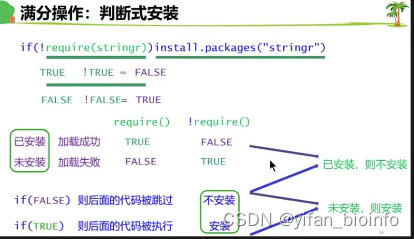
- 问题
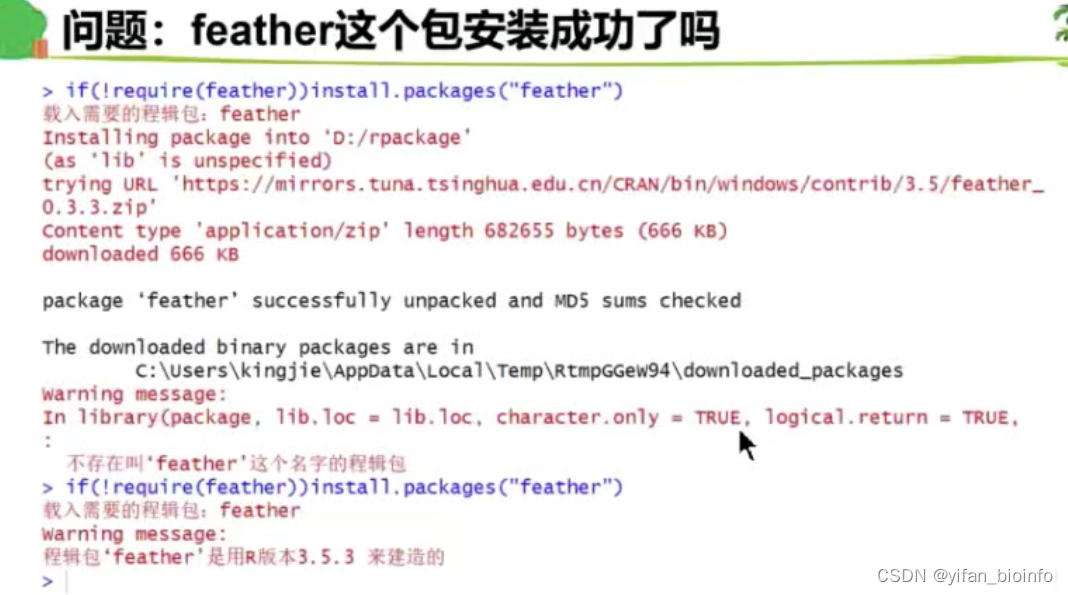
成功!
3.3.3R包的使用场景

3.3.4R包如何使用--获取帮助

如果你想自主学习一个R包,一个很好的办法就是先运行作者提供的Examples,通过控制台返回的结果,去分析R包的含义和使用方法。
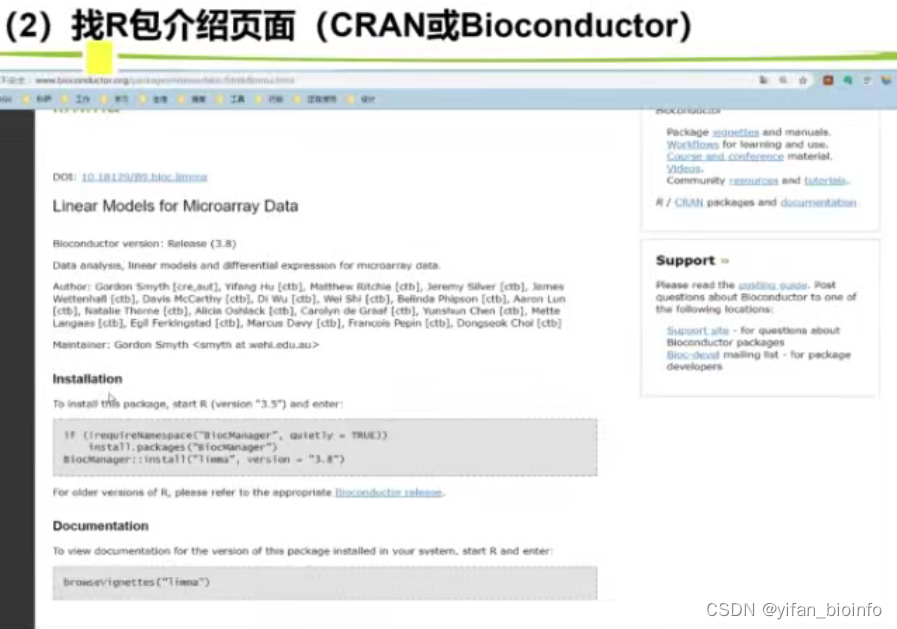
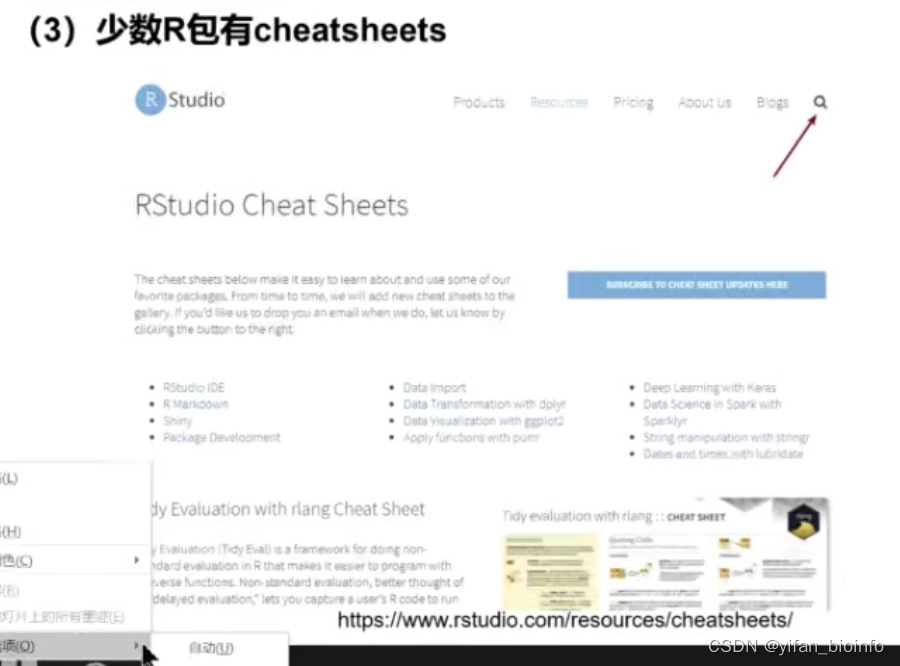

这就类似于使用小抄,除官方提供外,个人用户写的慎重判断使用
突发奇想地一个计算机领域知识小提示(这些通识类的知识对于一个资深的码农来说或许是不重要的,但如果你只是一个初学者,这对你逻辑思维体系的建立是很有帮助的,不要小瞧文字传达的逻辑性):
在计算机领域中,镜像通常指的是一个与另一个对象相似的复制品。具体来说,它可以有以下几种含义:
-
操作系统镜像: 在操作系统中,镜像是一个完整的、可启动的操作系统副本,包括所有的文件、配置和设置。通过创建操作系统镜像,可以轻松地在多台计算机上部署相同的操作系统环境。
-
软件镜像: 软件镜像是一个包含了软件及其依赖关系的完整副本。它通常用于分发软件,特别是对于大型软件或跨网络分发的软件而言,可以提高下载速度和减少服务器负载。
-
数据镜像: 数据镜像是一个包含了数据及其相关设置和结构的完整副本。这种镜像常用于数据备份、数据恢复、数据分发等应用。
-
镜像站点: 镜像站点是一个服务器,它存储了其他服务器上的数据、文件或软件的副本。镜像站点的存在可以提高数据、文件或软件的可用性和下载速度,尤其是对于广泛使用的内容而言。
在软件开发和分发中,镜像通常指的是软件镜像,它是一个包含了软件及其依赖关系的完整副本,用于快速分发和部署软件。
3.3.5R语言里的符号
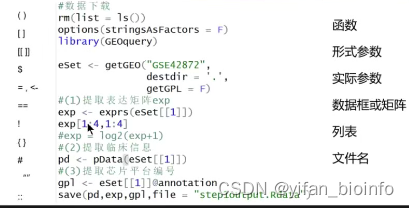
R语言里的符号:
()前面是 函数的名字
[] 是用来取子集的,如果里面没有逗号就是在对一个向量取子集,如果有逗号就是在对矩阵或者数据框取子集
[[]] 对列表取子集
$ 可以是数据框取一列,也可以是列表取一个元素
=,<- 这两者都可以表示赋值的含义,等号也可以表示形式参数和实际参数的连接。
== 判断是否相等,返回逻辑值
! True变False ,False变True
{} 里面都是代码
# 注释
'''' '' 两者都在表示字符串
:: 前面是包的名字,后面是函数的名字
四.文件读写
4.1文件读取
- csv文件
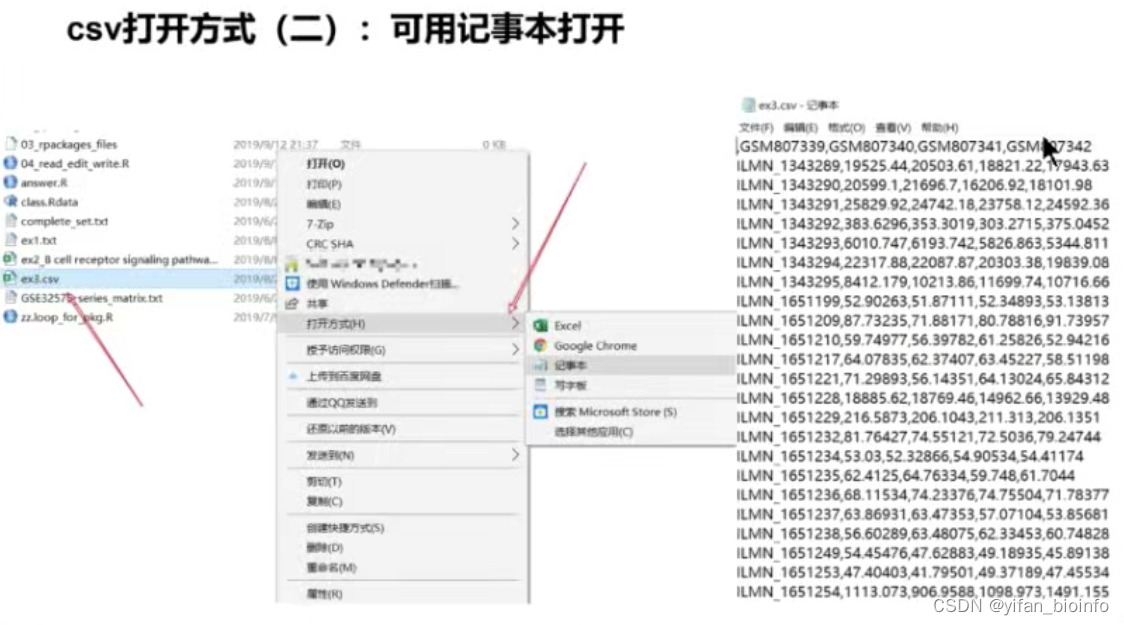
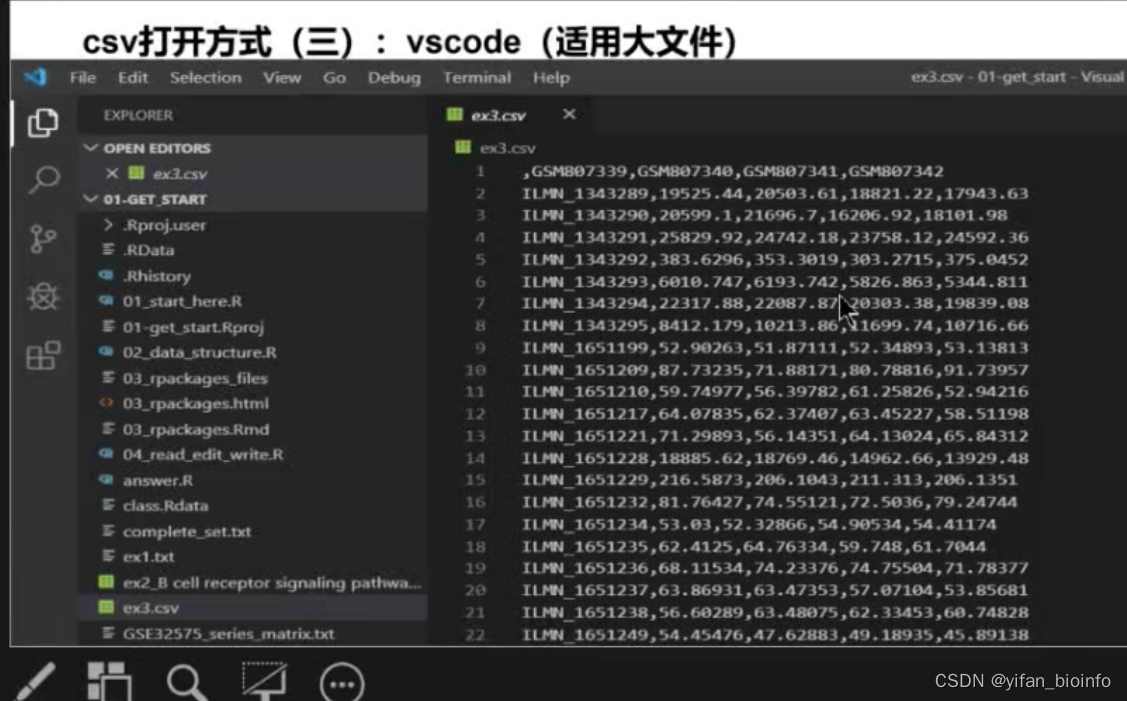
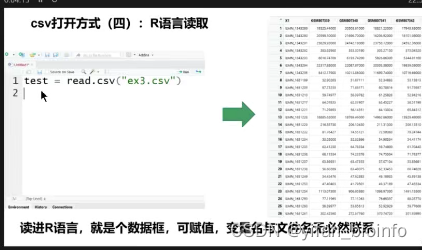
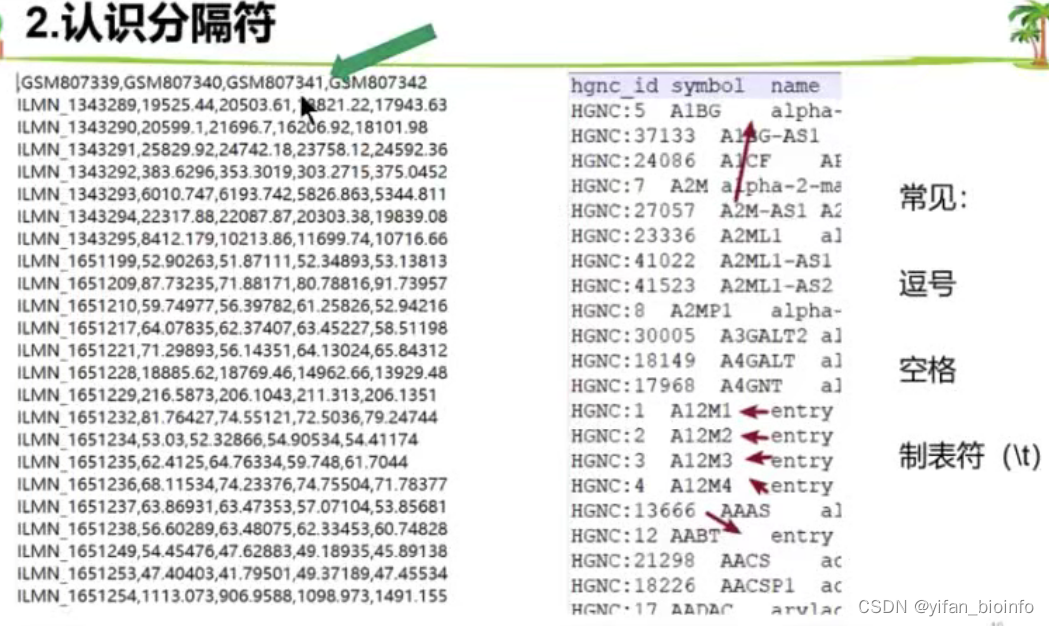

CSV :逗号分隔值 Tab:Tab分隔值
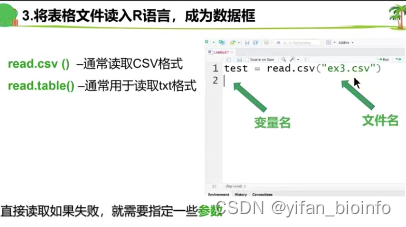
导出文件就是另存为,自己取名字!(理清这个逻辑)
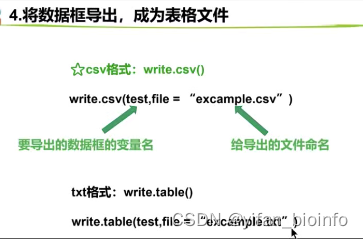

- 认识所有R文件

- 常见报错

- 练习
#文件读写部分代码
#1.读取ex1.txt
ex1 <- read.table("ex1.txt",header = T)
ex1
#2.读取ex2_B cell receptor signaling pathway.csv
ex2 <- read.csv("ex2_B cell receptor signaling pathway.csv",row.names = 1)
#3.读取GSE32575_series_matrix.txt
gse <- read.table("GSE32575_series_matrix.txt",
comment.char = "!",
header = T,
row.names = 1)
head(gse)
#4.描述gse的属性
dim(gse)
colnames(gse)
rownames(gse)
class(gse)
#5.将gse导出为txt。
write.table(gse,file = "gse.txt")
#6.将gse导出为csv文件
write.csv(gse,file = "gse.csv")
#7.将gse保存为Rdata并加载。
save(gse,file = "gse.Rdata")
rm(list = ls())
load(file = "gse.Rdata")
#1.fasta----
#BiocManager::install('seqinr')
library(seqinr)
library(stringr)
fasta <- read.fasta(file = "longreads.fa",
as.string = TRUE,
forceDNAtolower = FALSE)
#2.fastq----
rm(list = ls())
#BiocManager::install("ShortRead")
library(ShortRead)
rfq <- readFastq("s_1_sequence.txt")
rfq
sread(rfq)
id(rfq)
quality(rfq)
file <- tempfile()
writeFastq(rfq, file)
readLines(file, 8)
#3.vcf----
rm(list = ls())
#install.packages("vcfR")
library(vcfR)
vcf<-read.vcfR("pinf_sc50.vcf.gz")
class(vcf@meta)
class(vcf@fix)
class(vcf@gt)
meta = vcf@meta
str_length(vcf@meta)
fix = vcf@fix
fix[1:4,1:4]
gt = vcf@gt
colnames(gt)
gt[1:4,1:4]
#4.bam----
rm(list = ls())
suppressMessages(library(Rsamtools))
bamFile="wes.bam"
bam <- scanBam(bamFile)
names(bam[[1]])
bam=as.data.frame(do.call(cbind,lapply(bam[[1]],as.character)))
#5.bed----
rm(list = ls())
#BiocManager::install("genomation")
library(genomation)
refseq = readBed("chr21.refseq.hg19.bed",track.line=FALSE,remove.unusual=FALSE)
#6.gtf----
rm(list = ls())
gtf = data.table::fread("ANXA1.gencode.v7.gtf",data.table = F)
#7.gff---
rm(list = ls())
gff = data.table::fread("pinf_sc50.gff")
- 练习
看到了R_Day4_P2_readfile2.mov


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









