








–https://doi.org/10.1038/s41592-024-02305-7
模型代码详情:https://github.com/biomap-research/scFoundation
留意更多内容,欢迎关注微信公众号:组学之心
1.模型预训练
1.1 预训练数据收集和处理
1.1.1 数据收集
人的 scRNA-seq 数据存放在
数据主要从GEO、HCA、Single Cell Portal、EMBL-EBI、hECA、DISCO中获取。每个数据集都有链接到已发表的论文,具有相应的论文DOI ID。本研究从这些数据库中人工收集数据,并删除具有重复ID的数据集。
数据集包含健康人、各种疾病、各种癌症类型的各种组织的样本,有的 5000 多万个单细胞,代表了人类单细胞转录组的全谱。研究将所有数据分为训练数据集和验证数据集,验证数据集随机抽样得到,包含 10万个单细胞。具体的数据集名称和论文信息作者在supplementary files Data1和2中提供了。
-
1.大多数数据集含有raw count矩阵
-
2.对于标准化后的数据集,将它们转换回raw count形式(将raw count中最小的非零值视为1,将所有剩余的非零值除以这个最小值并取整数部分)。
-
3.对于TPM或FKPM数据集无法转换回raw count的数据集不做处理,能转换的就转换回raw count形式。
1.1.2 统一基因symbol名称
用HUGO提供的参考基因名称统一基因表达矩阵的基因名称。纳入的基因包含人类蛋白质编码基因和常见的线粒体基因,共计 19,264 个。用零值填充某些数据缺少的基因。HUGO网站:https://www.genenames.org/
1.1.3 基本质控
保留表达超过 200 个基因的细胞,用Seurat和Scanpy来操作。
1.2 embedding之前的处理(输入优化)
为什么要优化输入数据
- 单细胞数据中90%的数据是0(稀疏性),直接计算注意力会有很多冗余计算。
- 许多基因表现出低 UMI count,将 scRNA-seq 数据视为图像并利用卷积神经网络提取特征是不可行的,因为它为稀疏位置引入了大量冗余计算。
做法:分层贝叶斯下采样
分层贝叶斯下采样能够降低冗杂和噪音,增加模型鲁棒性
第一层:
引入了一个伯努利分布的随机变量 γ γ γ 来决定是否对 X X X 进行下采样。如果 γ = 0 γ = 0 γ=0,输入向量 X input \mathbf{X}^{\text{input}} Xinput 就是直接复制 X X X ;如果 γ = 1 γ = 1 γ=1,输入向量 X input \mathbf{X}^{\text{input}} Xinput 就是第二层给定的 X X X 的下采样数据。基因表达total count低于1000则它的质量不适合下采样,固定 γ = 0 γ = 0 γ=0。
第二层:
利用二项分布对每个基因的raw count数据进行下采样:
X
i
input
∼
B
(
X
i
,
b
)
X_{i}^{\text{input}} \sim B(X_i, b)
Xiinput∼B(Xi,b)
其中
X
i
input
X_{i}^{\text{input}}
Xiinput 是基因
i
i
i 的下采样raw count值,
X
i
X_{i}
Xi 是基因
i
i
i 的观察原始计数值,
b
b
b 是下采样率。对于每个细胞,所有基因共享相同的下采样率
b
b
b,因此,下采样后细胞的total count的期望满足:
E
[
T
C
(
X
input
)
]
=
E
[
X
1
input
+
⋯
+
X
N
input
]
=
E
[
X
1
input
]
+
⋯
+
E
[
X
N
input
]
=
b
⋅
X
1
+
⋯
+
b
⋅
X
N
=
b
⋅
T
C
(
X
)
E[TC(\mathbf{X}^{\text{input}})] = E[X_1^{\text{input}} + \cdots + X_N^{\text{input}}] = E[X_1^{\text{input}}] + \cdots + E[X_N^{\text{input}}] = b \cdot X_1 + \cdots + b \cdot X_N = b \cdot TC(\mathbf{X})
E[TC(Xinput)]=E[X1input+⋯+XNinput]=E[X1input]+⋯+E[XNinput]=b⋅X1+⋯+b⋅XN=b⋅TC(X)
其中 T C ( X ) TC(\mathbf{X}) TC(X)是原始细胞的 total count, T C ( X input ) TC(\mathbf{X}^{\text{input}}) TC(Xinput)是下采样细胞的total count。
这种设计确保了原始细胞和下采样细胞的总计数之比的期望固定为 1 / b 1/b 1/b,意味着在整个细胞样本中,经过下采样处理后,每个细胞的total count(基因表达值的总和)的期望值是原始细胞total count的 b b b 倍的倒数。但对于 X input X^{\text{input}} Xinput 中的每个基因,其表达值是随机变量的观测值。这种先验分布保证了在训练过程中可以看到输入和输出之间的不同倍数变化。
进一步让参数
b
b
b 服从Beta分布:
b
∼
Beta
(
2
,
2
)
b \sim \text{Beta}(2, 2)
b∼Beta(2,2)
-
Beta(2, 2)分布是一种Beta分布的特殊形式,属于连续概率分布,定义在区间 [0, 1] 上。Beta分布通常用于描述概率值的分布,例如概率参数 𝑝 的不确定性。用来归一化概率密度,使得总概率为1。
-
这种分布的概率密度函数表现为一个对称的钟形曲线,意味着它倾向于在中间值附近(即 0.5 附近)取值。这种分布表示对 𝑥 的均匀偏好,即 𝑥 既不太大也不太小,呈现中等的不确定性。(均值=0.5,方差=0.05)使得它可以表达对某个事件发生的概率没有强烈的偏好,但也不认为其非常小或非常大。
因此,输入基因表达向量
X
input
X^{\text{input}}
Xinput 可以定义为:
X
input
=
{
X
if
γ
=
0
[
B
(
X
1
,
b
)
,
B
(
X
2
,
b
)
,
…
,
B
(
X
N
,
b
)
]
if
γ
=
1
\mathbf{X}^{\text{input}} = \begin{cases} \mathbf{X} & \text{if } \gamma = 0 \\ [B(X_1, b), B(X_2, b), \ldots, B(X_N, b)] & \text{if } \gamma = 1 \end{cases}
Xinput={X[B(X1,b),B(X2,b),…,B(XN,b)]if γ=0if γ=1
γ ∼ Bernoulli ( 0.5 ) 且 b ∼ Beta ( 2 , 2 ) \gamma \sim \text{Bernoulli}(0.5) \quad \text{且} \quad b \sim \text{Beta}(2, 2) γ∼Bernoulli(0.5)且b∼Beta(2,2)
然后这个输入向量将被随机掩码并输入到 xTrimoGene 模型中。
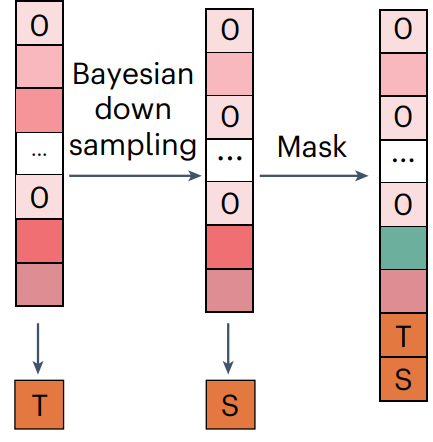
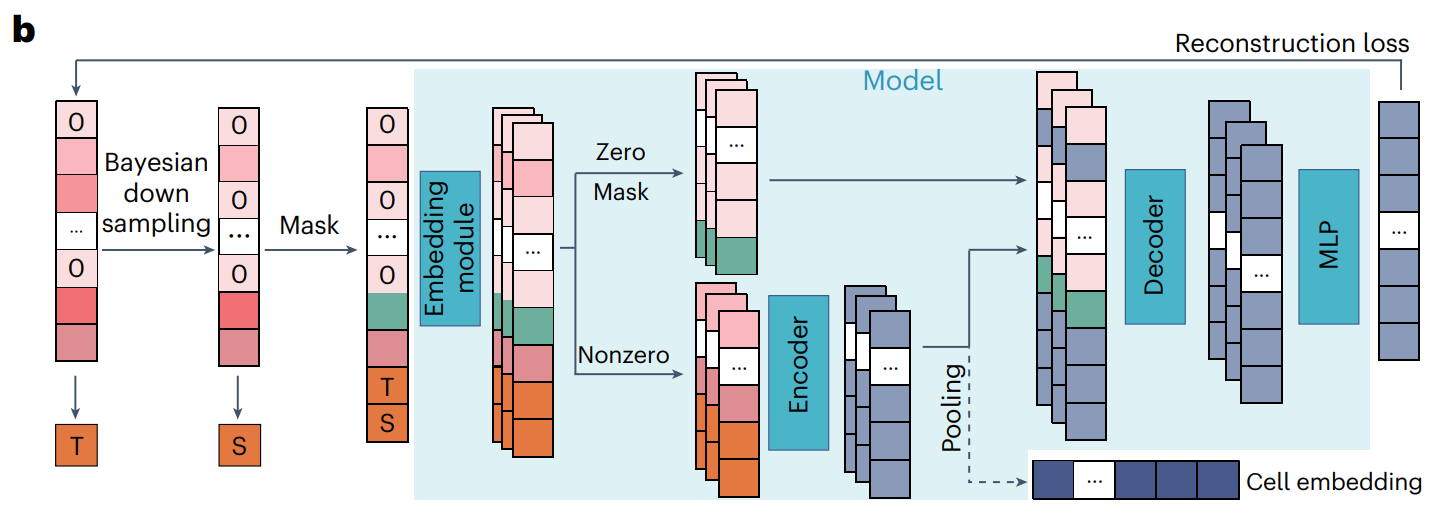
简单来说
左边一列是单个细胞的原始raw count 矩阵,先计算一个基因表达值的和“T”,然后每个基因贝叶斯下采样后得到中间一列,并计算一个下采样后的基因表达值的和“S”,最后对这个下采样的结果进行随机masked(0值部分随机masked 3%的,非0值有30%),拼上“T”和“S”,组成一个19266长度的向量,送到xTrimoGene模型中。
1.3 输入Embedding
- 学习的离散化方法或可学习的表示,是保留基因表达值的连续语义的理想选择。scBert将基因表达值四舍五入为整数值,这限制了模型区分基因表达值之间接近和相似性的能力。1.99 和 2.01 映射到 1 和 2,1.99 和 1.01 映射到 1。
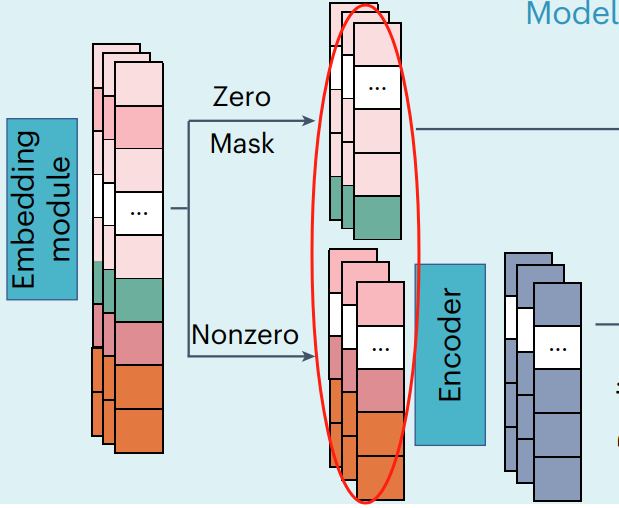
0和masked的值
直接随机初始化embedding
非0的值–自离散化映射
对于非零表达值,查找表
T
∈
R
b
×
d
T \in \mathbb{R}^{b \times d}
T∈Rb×d 被随机初始化,其中
d
d
d 是隐藏embedding维度,
b
b
b 是预定义的检索token数,研究中
b
=
100
b = 100
b=100。然后对于基因
i
i
i 的非零表达值 $x_i^{\text{input}}$ ,它首先被送入包含leaky ReLU激活的线性层,得到一个中间embedding
v
1
∈
R
b
\mathbf{v}_1 \in \mathbb{R}^{b}
v1∈Rb :
v
1
=
leakyReLU
(
x
i
input
×
w
1
)
\mathbf{v}_1 = \text{leakyReLU}(x_i^{\text{input}} \times \mathbf{w}_1)
v1=leakyReLU(xiinput×w1)
其中
w
1
∈
R
b
\mathbf{w}_1 \in \mathbb{R}^{b}
w1∈Rb 是参数。然后另一个线性层和一个缩放因子 𝛼 被用来进一步处理
v
1
{v}_1
v1:
v
2
=
v
1
⋅
w
2
+
α
×
v
1
\mathbf{v}_2 = \mathbf{v}_1 \cdot \mathbf{w}_2 + \alpha \times \mathbf{v}_1
v2=v1⋅w2+α×v1
其中
w
2
∈
R
b
×
b
\mathbf{w}_2 \in \mathbb{R}^{b × b}
w2∈Rb×b 和 𝛼 是可学习参数。得到的向量
v
2
∈
R
b
\mathbf{v}_2 \in \mathbb{R}^{b}
v2∈Rb 使用SoftMax函数进行归一化,得到注意力得分向量
v
3
∈
R
b
\mathbf{v}_3 \in \mathbb{R}^{b}
v3∈Rb:
v
3
=
[
v
3
0
,
…
,
v
3
i
,
…
]
v
3
i
=
exp
(
v
2
i
)
∑
i
=
1
b
exp
(
v
2
i
)
\mathbf{v}_3 = [v_{3_{0}}, \ldots, v_{3_{i}}, \ldots] \quad v_{3_{i}} = \frac{\exp(v_{2_{i}})}{\sum_{i=1}^b \exp(v_{2_{i}})}
v3=[v30,…,v3i,…]v3i=∑i=1bexp(v2i)exp(v2i)
基因
i
i
i 的最终embedding
E
i
∈
R
d
\mathbf{E}_i \in \mathbb{R}^{d}
Ei∈Rd 是查找表
T
T
T 中所有embedding的加权和:
E
i
=
v
3
⋅
T
E_i = \mathbf{v}_3 \cdot T
Ei=v3⋅T
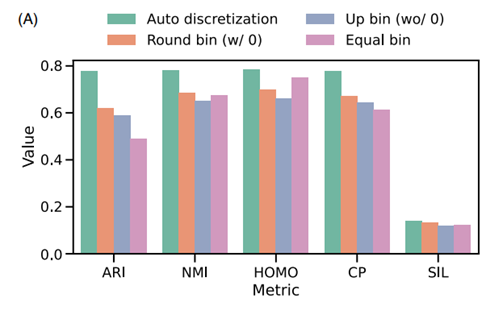
离散化方法效果的比较
研究将所提出的自离散化策略的性能与其他三种离散化方法进行了比较:
- (1) 将 bin 舍入为零,其中值四舍五入到最接近的整数,零保持原样
- (2) 没有零的 Up bin。大于 0 的值将转换为最接近的上限整数,而 0 表示为单个 0。
- (3) 相等的 bin。所有值都属于一个固定的百分比区间,该区间由值分布和频率计算得出。
研究在标准细胞聚类任务上评估了不同的策略,发现所提出的自离散化策略优于其他策略,证明了高分辨率投影在处理表达值中的重要性。
基因表达值embedding+基因ID embedding
刚刚都是对单个细胞而言,而对完整的数据来说还需要其它的一些操作,图是xTrimoGene论文中的,这里简单描述一下:
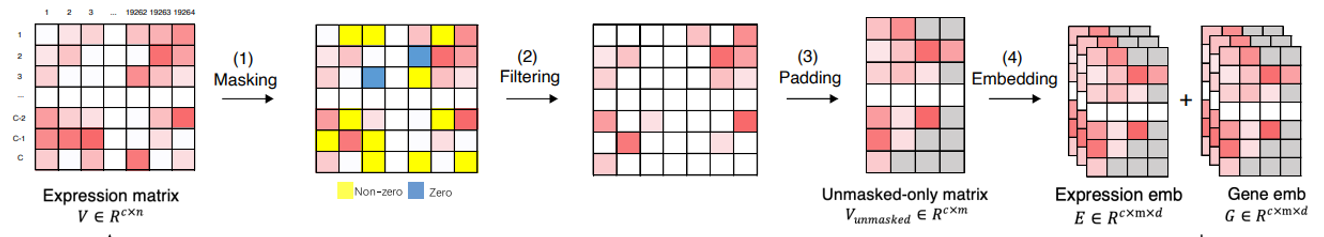
- Filtering:在原本矩阵中去掉masked的部分(用0填上)
- Padding:这里一行是一个细胞,把细胞各自有的基因挑出来放一起(红色部分),这时候比如一个细胞有A基因,但是另一个细胞没有,那就padding填充上去[pad]特殊字符(灰色部分),填充后大家集体有的基因数量则为m,图中c是细胞数量,d是embedding的维度。
- Embedding:就是上面提到的自离散化映射,再加上ID的embedding,组成输入embedding。
1.4 Encoder
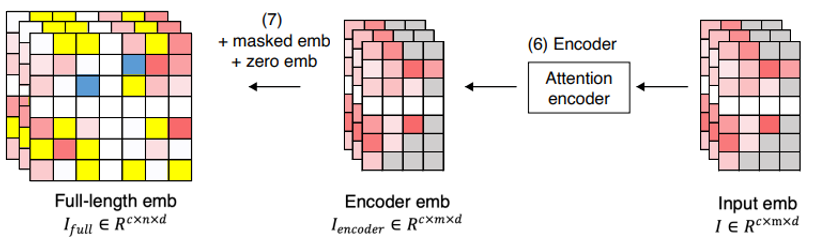
encoder设计为仅关注未masked矩阵的非零部分 V u n m a s k e d V_{unmasked} Vunmasked。该编码器基于传统的多头注意力transformer,并将值嵌入 E 和基因嵌入 G 的组合作为其输入 $I ∈ R^{c×m×d} $。
值嵌入和基因嵌入分别类似于自然语言建模中的单词和位置嵌入。值嵌入 E 是使用自离散化策略生成的,而基因嵌入 G 是从函数 f L f_L fL 中检索的,该函数将基因符号映射到嵌入的词汇表中。
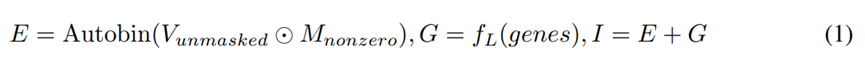
然后,encoder处理输入嵌入 I I I,并通过多头注意力机制生成高级基因表示 I e n c o d e r ∈ R c × m × d I_{encoder} ∈ R^{c×m×d} Iencoder∈Rc×m×d
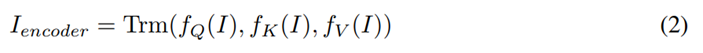
Trm 表示 Transformer 块。Encoder只对基因子集进行操作,将处理序列的长度减少到原始序列的 1/10。这允许在没有任何计算近似的情况下使用全长 transformer。
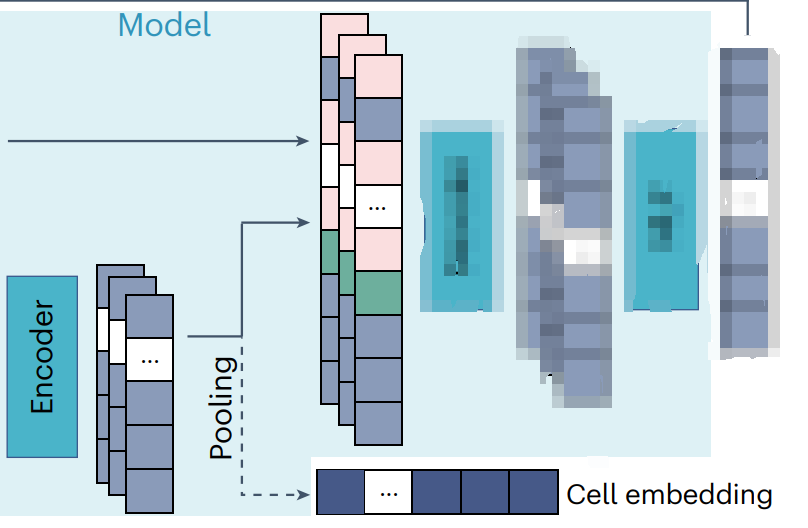
Encoder的输出一个是和0值+masked的随机embedding拼接起来,作为后续decoder的输入 I f u l l I_{full} Ifull;另一方面是做个pooling操作生成cell embedding,用于细胞级下游任务。
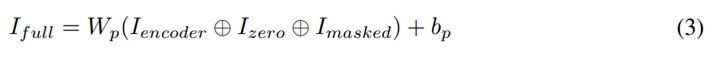
1.5 Decoder
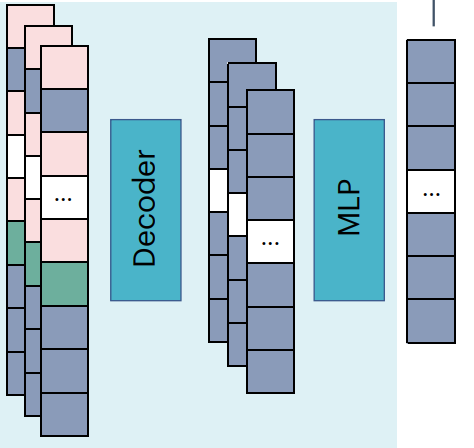
Decoder在转录组范围内恢复表达值,包括零表达和掩码位置的基因;并预测基因的表达水平。
研究使用基于核的近似变体Performer作为decoder的主干,因为对于长序列,注意力计算很困难。在Performer中,使用了核化的注意力机制,近似计算注意力权重:
Att
‾
(
Q
,
K
,
V
)
=
D
^
−
1
(
∅
(
Q
)
(
∅
(
K
)
T
V
)
)
D
^
−
1
=
diag
(
∅
(
Q
)
(
∅
(
K
)
)
T
1
K
)
\overline{\text{Att}}(Q, K, V) = \hat{D}^{-1} \left( \varnothing(Q) \left( \varnothing(K)^T V \right) \right) \hat{D}^{-1} = \text{diag} \left( \varnothing(Q) \left( \varnothing(K) \right)^T \mathbf{1}_K \right)
Att(Q,K,V)=D^−1(∅(Q)(∅(K)TV))D^−1=diag(∅(Q)(∅(K))T1K)
可核化注意力机制通过引入核技巧,将复杂度降低到线性级别。其中 ∅ ( ⋅ ) \varnothing(·) ∅(⋅) 是用于近似原始注意力方程中矩阵 A A A 的核函数。
decoder的输出是
χ
Out
\chi^{\text{Out}}
χOut:
χ
Out
=
Performer
(
χ
Dec-input
)
∈
R
19
,
266
×
f
\chi^{\text{Out}} = \text{Performer}(\chi^{\text{Dec-input}}) \in \mathbb{R}^{19,266 \times f}
χOut=Performer(χDec-input)∈R19,266×f
1.6 MLP预测基因表达值
为了预测表达值,去掉了
T
T
T 和
S
S
S 的embeddings,并且使用多层感知器(MLP)将
χ
Out
\chi^{\text{Out}}
χOut 投射为标量。这些标量形成了预测向量
P
P
P:
P
=
MLP
(
χ
Out
)
∈
R
19
,
264
\mathbf{P} = \text{MLP}(\chi^{\text{Out}}) \in \mathbb{R}^{19,264}
P=MLP(χOut)∈R19,264
所有参数 Θ = { E i , T i G , Θ Encoder , Θ Decoder , Θ MLP } \Theta = \{\mathbf{E}_i, \mathbf{T}_i^G, \Theta_{\text{Encoder}}, \Theta_{\text{Decoder}}, \Theta_{\text{MLP}}\} Θ={Ei,TiG,ΘEncoder,ΘDecoder,ΘMLP} 在预训练期间进行了优化。
1.7 Mean square error (MSE) loss
作者使用回归损失作为损失函数:
L
=
1
∣
M
∣
∑
i
=
0
∣
M
∣
(
X
i
−
P
i
)
2
L = \frac{1}{|M|} \sum_{i=0}^{|M|} (X_i - P_i)^2
L=∣M∣1i=0∑∣M∣(Xi−Pi)2
其中,
M
M
M 是一个二进制向量,指示哪些基因被掩码(值为1表示被掩码),
X
i
X_i
Xi 和
P
i
P_i
Pi 分别是真实值和预测的基因表达值。|| 是1-范数运算符。
选择性只对被掩码的基因计算损失,从而关注那些有预测需求的基因表达值。通过将总损失除以掩码基因的数量( 1 ∣ M ∣ \frac{1}{|M|} ∣M∣1),实现对不同数量掩码基因的归一化,从而使损失值具有可比性。
1.8 encoder和decoder的设计
与encoder相比,decoder的模型尺寸更小,嵌入尺寸更小,注意力层更少,注意力头更少。例如,在最大的模型配置中,encoder和decoder之间的层深比为 2:1,头数比为 1.5:1。同样,非对称encoder-decoder设计的原理已在masked自动编码器 (MAE)中被证明是强大的,该算法是为 CV 数据预训练量身定制的。
与 MAE 不同,xTrimoGene 利用偏向masked策略来避免学习过程被零标记所主导。尽管 scRNA-seq 数据与图像不同,但研究的结果表明,xTrimoGene 的性能增益与 MAE 相当,具有更高效的训练和更好的下游任务性能。
2.使用scFoundation
2.1 准备工作
配置环境
模型的代码在:https://github.com/biomap-research/scFoundation
git clone git@github.com:biomap-research/scFoundation.git
推荐用:python3.9.19,cuda11.8,scanpy1.10.2,torch2.0.1
模型权重
–https://hopebio2020-my.sharepoint.com/:f:/g/personal/dongsheng_biomap_com/Eh22AX78_AVDv6k6v4TZDikBXt33gaWXaz27U9b1SldgbA
数据使用
–https://doi.org/10.6084/m9.figshare.24049200
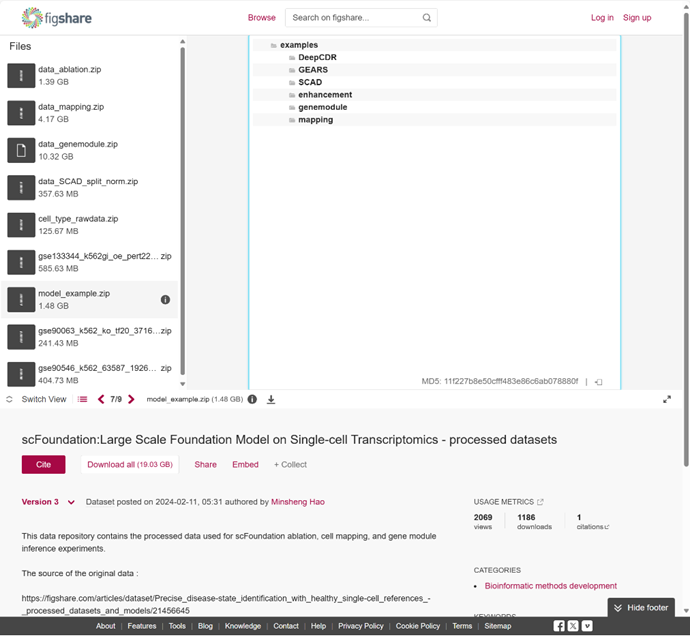
用到其中的model_example数据,解压后该文件夹叫example
2.2 Inference
统一基因名称
要把数据的gene symbol和作者提供的基因list匹配上。
–scFoundation/model/OS_scRNA_gene_index.19264.tsv
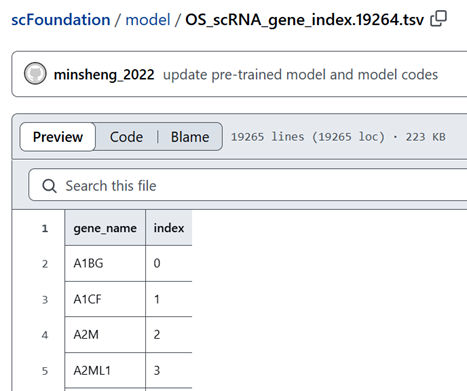
可以通过scFoundation/model/get_embedding.py
gene_list_df = pd.read_csv('../OS_scRNA_gene_index.19264.tsv', header=0, delimiter='\t')
gene_list = list(gene_list_df['gene_name'])
X_df, to_fill_columns, var = main_gene_selection(X_df, gene_list)
然后把X_df保存成npy或者csv格式
获取细胞embedding
在 scFoundation/model/demo.sh 文件中,作者提供了几个脚本来推断各种类型的嵌入,包括单细胞、批量和基因嵌入。要运行这些脚本,只需复制相应的 Python 命令并将其粘贴到命令行或终端中。
这里是推断cell embedding的示例命令,用的是example中的Baron_enhancement.csv:
### Cell embedding
python get_embedding.py --task_name Baron --input_type singlecell --output_type cell --pool_type all --tgthighres a5 --data_path ./examples/enhancement/Baron_enhancement.csv --save_path ./examples/enhancement/ --pre_normalized F --version rde
运行脚本后,模型会自动加载预训练好的权重,生成的嵌入examples/enhancement/Baron_01B-resolution_singlecell_cell_embedding_a5_resolution.npy可以用于文章的下游任务分析。
获取基因embedding
### Cell embedding
python get_embedding.py --task_name Baron --input_type singlecell --output_type gene --pool_type all --tgthighres a5 --data_path ./examples/enhancement/Baron_enhancement.csv --save_path ./examples/enhancement/ --pre_normalized F --version rde
参数调整
为了快速启动,只需修改 --data_path 以指向自己的数据。以下是每个参数的详细描述:
-
input_type:指定输入的类型。singlecell,bulk,默认是singlecell
-
output_type: 确定输出的类型。cell,gene,gene_batch,默认是cell
注意:在 cell 模式下,输出形状为 (N,h),其中 N 是细胞数量,h 是隐藏维度。在 gene* 模式下,输出形状为 (N,19264,h),其中 N 是细胞数量,19264 是基因数量,h 是隐藏维度。在 gene 模式下,每个细胞的基因嵌入都是单独处理的。在 gene_batch 模式下,数据中的所有细胞都被视为单个批次并一起处理。确保在此模式下输入细胞的数量不超过 5。 -
pool_type:定义细胞嵌入的池类型。all,max,默认是all
获取 cell embeddings 的方法,仅当 output_type 设置为 cell 时才可用。 -
tgthighres:设置T token的值。默认是t4
注意:可以设置三种方式 - 目标高分辨率,即 T=number(以 ‘t’ 开头)、高分辨率倍数变化,即 T/S=number(以 ‘f’ 开头)或高分辨率附加T=S+number(以 ‘a’ 开头)。仅当 input_type 为 singlecell 时有效。 -
pre_normalized:控制 S token的计算方法。F, T, A,默认是F
注意:当 input_type 为 singlecell 时,T 或 F 表示输入基因表达数据是否已标准化 +log1p。A 表示数据已标准化 +log1p,并在末尾附加总计数,从而得到 N*19265 的数据形状。此模式用于 GEARS 任务。对于批量输入类型,F 表示 T 和 S 标记值为 log10(基因表达的总和),而 T 表示它们是未经对数变换的总和。这对于具有少量测序基因的批量数据很有用。 -
version: Model versions for generating cell embeddings.默认是ce
注意:读取深度增强时使用 rde,其他情况使用 ce。仅当 output_type 为 cell 时有效。
2.3 微调
要微调或将 scFoundation 模型与其他层或模型集成,可以参考 finetune_model.py 文件中提供的示例模型代码。
代码中建议frozen token_emb、pos_emb和encoder除了倒数第二层的参数,encoder输出得到logits,对logits最大池化后得到每个样本的最大特征,随后再做归一化(self.norm = torch.nn.BatchNorm1d(model_config[‘encoder’][‘hidden_dim’], affine=False, eps=1e-6)),并输入到2层全连接层,输出最终的分类,可以用于细胞类型注释。
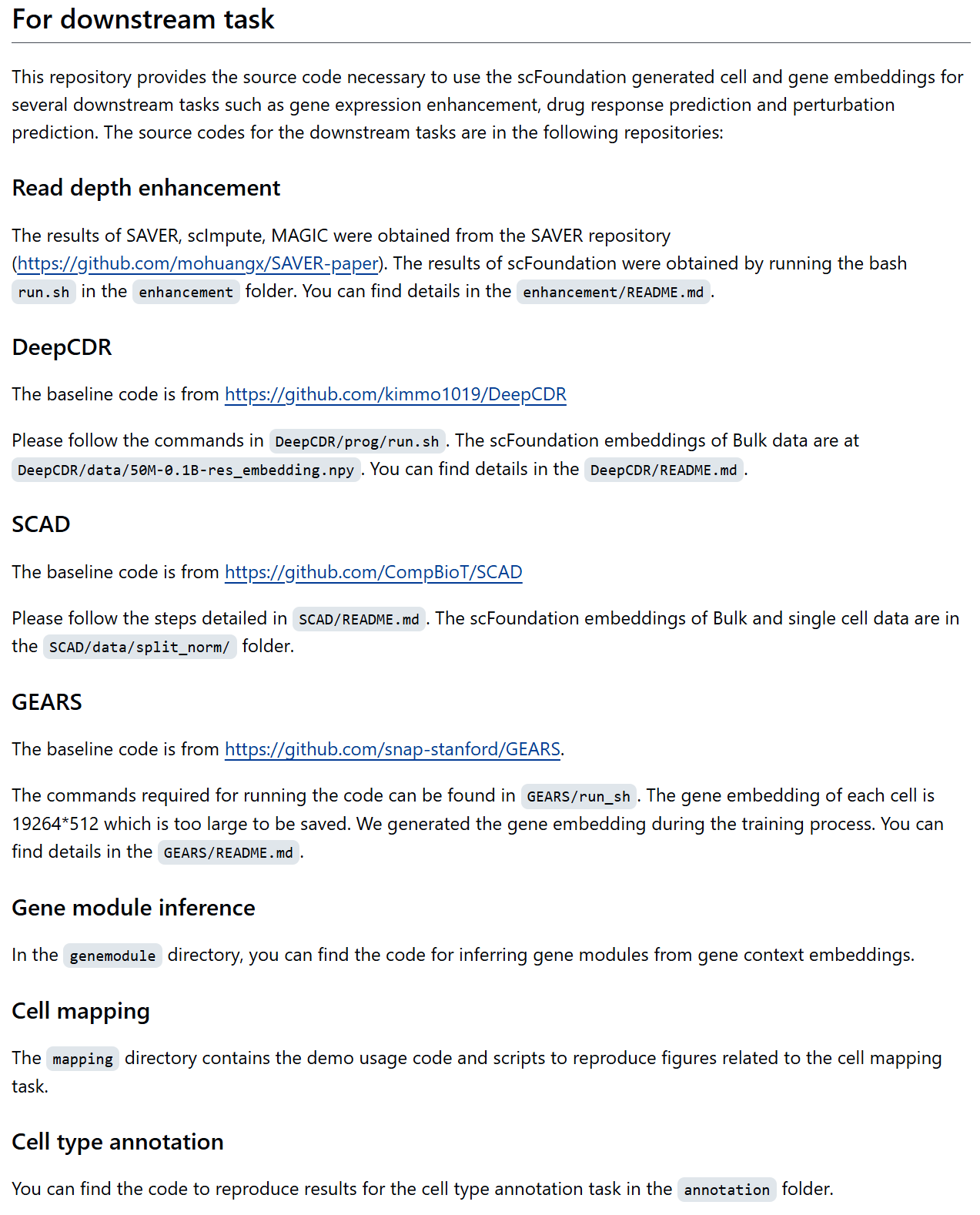
下游任务




















 1698
1698

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











