







本文为Coursera课程 Assembling Genomes and Sequencing Antibiotics (Bioinformatics II) 笔记。
作者:ybw
Introduction
Motivation
A failure attempt - Finding antibiotics from genome
因为抗生素为由氨基酸(amino acid)组成的序列,根据生物学中心法则,DNA产生RNA,RNA产生蛋白质。
The Central Dogma of Molecular Biology states that “DNA makes RNA makes protein.”
因此,首先可以想到将抗生素反编译为DNA序列,并在微生物的DNA中寻找可能产生该抗生素的DNA子串。
根据中心法则(如图1),
图1:Central dogma.
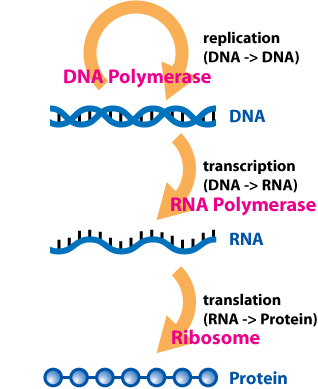
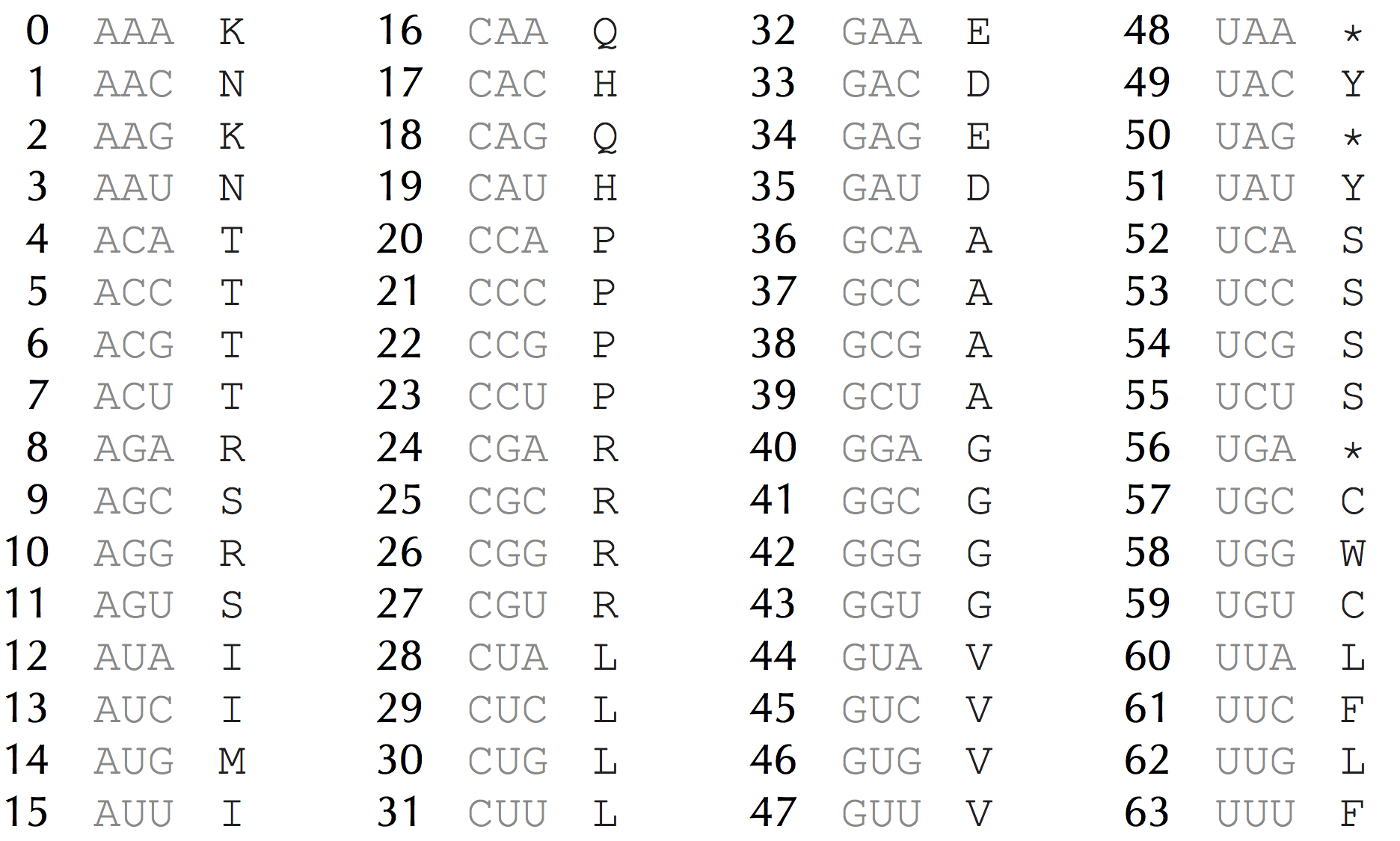
DNA首先转录(transcribe)单链RNA,RNA由{A, G, C, U}四个核糖核苷酸组成(four ribonucleotides: adenine, guanine, cytosine, and uracil);之后,RNA按一定规则翻译(translate)为蛋白质的氨基酸序列。这个规则即每3个RNA核苷酸组成一个密码子(codon),每个密码子翻译为一种氨基酸。所有密码子一共可翻译为20种不同的氨基酸,外加3个终止密码子(stop codons),如图2。
图2:Genetic code.
在实际中,抗生素多为环形多肽(cyclic peptides, cyclopeptide - 环肽),如图3所示为Tyrocidine B1所包含的10个氨基酸的顺序,可对应最多10中不同的氨基酸线性顺序。然而,即使穷举所有可能生成该环形多肽的DNA序列,也无法在产生Tyrocidine B1的Bacillus brevis(短芽孢杆菌)的基因中找到对应的序列。因为抗生素并非是由DNA转录翻译所得到的。
图3:Tyrocidine B1 is a cyclic peptide (left), and so it has ten different linear representations (right).

1963年,Edward Tatum(Nobel laureate )做了一个实验,他在实验中抑制了执行Bacillus brevis中从事蛋白质翻译工作的分子机器ribosome(核糖体),之后,他观察到Bacillus brevis体内的蛋白质合成停止了,但tyrocidines和gramicidins的合成却没有。由此,Tatum得到一个假说,即这些多肽一定是由某种非核糖体机制(non-ribosomal mechanism)所产生的。1969年,Fritz Lipmann (another Nobel laureate) 证明了该假说。他将tyrocidines和gramicidins这类多肽称为非核糖体肽(non-ribosomal peptides,NRPs),而这些多肽是由一种名为NRP synthetase(合成酶) 的巨大蛋白质合成的。
细菌和真菌使用NRPs来消灭它们的敌人,但NRPs并非只针对病原体(pathogens),它的功能还包括抗肿瘤(anti-tumor)、免疫抑制(immunosuppressors)以及用于细菌间的交流(quorum sensing)。
Quorum Sensing
课程中提到了一个有趣的现象,即quorum sensing。人们之前一直认为细菌都是独行侠,直到研究人员发现了quorum sensing现象。这项发现表明细菌在移居到一个营养充足的环境,或是生成生物膜(biofilm formation)来抵御恶劣环境时,都有相互协调活动的能力。而细菌之间交流的手段是通过交换多肽或者其他一些分子实现的,这些物质称为细菌的费洛蒙(bacterial pheromones)。所传达的信息既可以是善意的,也可以是恶意的(amicable or adversarial)。
单个细菌释放的bacterial pheromones微乎其微,而当细菌数量增加,释放的pheromone浓度达到某个阈值(threshold)之后,细菌就会对其作出反应,特定的基因会被激活。
The method of peptides analysis - mass spectrum
由于NRPs并不遵循Central Dogma,无法通过基因来分析其组成。因此,只能从NRPs本身入手。研究人员通过质谱分析(mass spectrum)来推断其组成。Mass spectrum是一种在分子规模将需要分析的分子打成碎片,并测量碎片质量的方法。单位是daltons (Da, 碳的同位素12C原子的质量为12dalton)。在实际中,由于质子和中子的质量不同,以及同位素的存在,分子的质量都不是整数,如glycine(甘氨酸)的质量约为57.02 Da。 这里为了简单,均使用整数来表示核苷酸的质量。图下标所示。
Integer mass table:
| G | A | S | P | V | T | C | I | L | N | D | K | Q | E | M | H | F | R | Y | W |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 57 | 71 | 87 | 97 | 99 | 101 | 103 | 113 | 113 | 114 | 115 | 128 | 128 | 129 | 131 | 137 | 147 | 156 | 163 | 186 |
Sequencing with theoretical spectrum
Subpeptides
假设mass spectrum可能打破peptides的任何两个相连的氨基酸之间的连接,那么最后可以得到一个peptide所有的线性片段(linear fragments),称之为subpeptides。注意,当原peptide中有重复片段时,subpeptides也会有重复。
如:
NQEL有12个subpeptides: N, Q, E, L, NQ, QE, EL, LN, NQE, QEL, ELN, LNQ;
ELEL也有12个subpeptides: E, L, E, L, EL, LE, EL, LE, ELE, LEL, ELE, LEL。
注意:环形和线性的subpeptides是不同的(环形包括更多的subpeptides)。
一个peptide的所有subpeptides的质量(mass),再加上0和peptide自身的mass,从小到大排成一列,即是一个peptide的theoretical spectrum。
The theoretical spectrum of a cyclic peptide Peptide, denoted Cyclospectrum(Peptide), is the collection of all of the masses of its subpeptides, in addition to the mass 0 and the mass of the entire peptide, with masses ordered from smallest to largest.
如,NQEL的theoretical spectrum如下。
| 0 | 113 | 114 | 128 | 129 | 227 | 242 | 242 | 257 | 355 | 356 | 370 | 371 | 484 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L | N | Q | E | LN | NQ | EL | QE | LNQ | ELN | QEL | NQE | NQEL |
Generating Theoretical Spectrum
如何产生一个环肽的theoretical spectrum? 下面给出两种方法。(index从1开始)
- 在peptide后加上相同的peptide,从i = 1 to |peptide|,分别提取长度为1 to |peptide|-1的子串;
- 课程给出的方法(只能求subpeptides的质量,不能求subpeptides的具体字母序列):
先求linear spectrum,当对应的subpeptides处于[1, |peptide|-1]之间时,用peptide的总质量减去该subpeptide的质量,即得到cyclopeptide中另一半subpeptide的质量。
如peptide = NQEL, subpeptide = QE,则 mass(LN)=mass(NQEL)−mass(QE) 。
Algo 1 pseudo-code:
CyclicSpectrum(Peptide, AminoAcid, AminoAcidMass)
doublePep ← Peptide + Peptide
CyclicSpectrum ← a list consisting of the single integer 0
for i ← 1 to |Peptide|
for j ← 1 to |Peptide|-1
// substring is from the i-th to (i+j)-th of the doublePep
subPep = doublePep(i,i+j)
add Mass(subPep) to CyclicSpectrum
add Mass(Peptide) to CyclicSpectrum
return sorted list CyclicSpectrumAlgo 1 python code:
def theoretical_spectrum(peptide):
double_pep = peptide * 2
cyclic_spectrum = [0]
for i in range(1,len(peptide)):
for j in range(len(peptide)):
subPep = double_pep[j:j+i]
m = mass( subPep )
cyclic_spectrum.append( m )
# add the mass of the whole peptide in the cyclic_spectrum
cyclic_spectrum.append( mass(peptide) )
cyclic_spectrum.sort()
return cyclic_spectrumAlgo 2 pseudo-code:
CyclicSpectrum(Peptide, AminoAcid, AminoAcidMass)
PrefixMass(0) ← 0
for i ← 1 to |Peptide|
for j ← 1 to 20
if AminoAcid(j) = i-th amino acid in Peptide
PrefixMass(i) ← PrefixMass(i − 1) + AminoAcidMass(j)
peptideMass ← PrefixMass(|Peptide|)
CyclicSpectrum ← a list consisting of the single integer 0
for i ← 0 to |Peptide| − 1
for j ← i + 1 to |Peptide|
add PrefixMass(j) − PrefixMass(i) to CyclicSpectrum
if i > 0 and j < |Peptide|
add peptideMass - (PrefixMass(j) − PrefixMass(i)) to CyclicSpectrum
return sorted list CyclicSpectrumAlgo 2 python code:
def cyclic_spectrum(pep):
"""
If Peptide represents a cyclic peptide.
与linear spectrum的不同之处: 减去中间部分的,即得到 subpeptides wrapping around the end of Peptide.
如:
Peptide = 'NQEL',
the mass of LN is equal to Mass(NQEL) minus the mass of QE, or 484 − 257 = 227.
"""
mass_table = { 'G': 57, 'A': 71, 'S': 87, 'P': 97, 'V': 99, 'T': 101, 'C': 103, 'I': 113, 'L': 113, 'N': 114, 'D': 115, 'K': 128, 'Q': 128, 'E': 129, 'M': 131, 'H': 137, 'F': 147, 'R': 156, 'Y': 163, 'W': 186 }
prifix_mass = [0] * (len(pep) + 1)
for i in range(1, len(pep)+1):
prifix_mass[i] = prifix_mass[i-1] + mass_table[pep[i-1]]
peptideMass = prifix_mass[-1]
cyclic_spec = [0]
for i in range(len(pep)):
for j in range(i+1, len(pep)+1):
cyclic_spec.append( prifix_mass[j] - prifix_mass[i] )
# the main difference from linear spectrum
if i > 0 and j < len(pep):
m = peptideMass - (prifix_mass[j] - prifix_mass[i])
cyclic_spec.append(m)
cyclic_spec.sort()
return cyclic_specFrom spectrum to peptides
The size of problem
假设组成peptides的氨基酸有如下18个不同的质量:
| G | A | S | P | V | T | C | I/L | N | D | K/Q | E | M | H | F | R | Y | W |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 57 | 71 | 87 | 97 | 99 | 101 | 103 | 113 | 114 | 115 | 128 | 129 | 131 | 137 | 147 | 156 | 163 | 186 |
- 对于一个给定的质量
M
,共有多少种不同的peptides符合该条件?
—— 使用动态规划方法解决该问题。
Optimal Substructure:
Let
Then,
S(M−57) is an optimal solution with respect to the peptides of S(M) taking of an amio acid G.- S(M−71) is an optimal solution with respect to the peptides taking of an amino acid A.
- ⋯
- S(M−186) is an optimal solution with respect to the peptides taking of an amino acid W.
Finally, S(M)=sum{S(M−57),⋯,S(M−186)}
Pseudo-code
Input : m
Output : n, The number of linear peptides having integer mass m.
Note : only focus on mass, assume that the mass 113 of **I/L** as one,
the same as mass 128 of **K/Q**.
S = an array of length m+1
S[0] = 1
S[j] = 0 for j = 1 to m
Mass = [57,71,87,97,99,101,103,113,114,115,128,129,131,137,147,156,163,186]
For i = 1 to m
For j = 0 to 17
if i >= Mass[j]
S[i] = S[i] + S[i - Mass[j]]
return S[m]Python code
def count_peptides_by_mass(m):
mass_list = \
[57,71,87,97,99,101,103,113,114,115,128,129,131,137,147,156,163,186]
count = [0] * (m+1)
count[0] = 1
for i in range(1,m+1):
count[i] = sum( [count[i-c] for c in mass_list if i >= c ] )
return count[-1]
经试验,令
N=S(m)
,则
N
与
A Branch-and-Bound Algorithm for Cyclopeptide Sequencing
由于只能通过brute force serach方法来推断peptides的组成,而问题规模又成指数型增长。因此需要使用branch-and-bound(分枝定界)算法来剪枝,从而减小问题规模,提高算法效率。
考虑从一个empty peptide(denoted as “”)来一步步(one amino acid by one)生成符合条件(给定的spectrum)的linear peptides。由于linear peptides的subpeptides是cyclic peptides的真子集(注意:但由于subpeptides中含有重复项,因此不能简单的使用python中的集合来判断),因此,在每次bounding时,要求生成的linear peptides一定要和给定的Spectrum相consistent。
Given an experimental spectrum
Spectrum of a cyclic peptide, a linear peptide is consistent with Spectrum if every mass in its theoretical spectrum is contained in Spectrum .下面给出算法。
The pseudocode for the branch-and-bound algorithm, called CYCLOPEPTIDESEQUENCING:CYCLOPEPTIDESEQUENCING(Spectrum) Peptides ← a set containing only the empty peptide while Peptides is nonempty Peptides ← Expand(Peptides) for each peptide Peptide in Peptides if Mass(Peptide) = ParentMass(Spectrum) if Cyclospectrum(Peptide) = Spectrum output Peptide remove Peptide from Peptides else if Peptide is not consistent with Spectrum remove Peptide from Peptides…待续















 479
479

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









