






目前该笔记还在编辑修改阶段,有部分内容未完善,请待后续更新
环境与工具准备
系统与硬件
Linux系统,建议具备交互式界面方便分析操作,至少一张Nvidia独立显卡。本篇笔记使用的是Win11的WSL2 Ubuntu 22.04.5 LTS,64G内存,4080s。
安装Gromacs (GPU-CUDA)
由于不同版本的Gromacs对于环境的依赖要求不同,这边不放出实际的安装命令,请根据Gromacs实际需求依赖版本安装环境
基础环境准备:gcc、 g++、 python、cmake、显卡驱动和CUDA(fuck Nvidia)
这边特别说明一下使用WSL2时,目前新的Nvidia驱动已经支持WSL2,也就是说不能在WSL2中安装任何Linux显卡驱动,这将会导致问题的出现,只需要安装CUDA Toolkit即可(必须是单独的工具包,因为默认的 CUDA 工具包附带一个驱动程序,并且很容易用默认安装覆盖 WSL 2 NVIDIA 驱动程序,官方下载链接:CUDA Toolkit 12.8 Update 1 Downloads | NVIDIA Developer)
更多细节请查看NVIDIA官方文档:1. NVIDIA GPU Accelerated Computing on WSL 2 — CUDA on WSL 12.8 documentation
Gromacs安装包:Welcome to the GROMACS documentation! — GROMACS documentation
安装完成后输入检测安装是否成功
gmx -version如果无法执行命令,先检查环境变量是否正确,以及更新修改后的环境变量
安装Amber24与AmberTools
官网下载地址:Download Amber MD
注意,通常情况只需要AmberTools即可完成对配体的拓扑化生成,无需安装完整Amber!
可以选择使用conda安装预编译的AmberTools或者下载源码编译
以下关于Amber的内容如果只安装AmberTools可不关注
Amber24 目前已经非商业/学术免费,填写姓名和机构,从官网获取Linux系统的源码,需要自己手动编译,建议使用Conda,mamba一类的包管理器进行安装,避免奇怪的环境依赖问题
这边本人参考的安装教程:https://zhuanlan.zhihu.com/p/666826404

可以编译的版本有CPU串行版(常规版)、CPU并行版、GPU串行版、GPU并行版,对于大部分用户来说选择常规版或GPU串行版即可
| 版本 | 适用对象 |
| CPU串行版(常规版) | 不需要CUDA、MPI,适用于无 NVIDIA GPU 或未安装驱动/CUDA的平台,也无需安装MPI |
| CPU并行版 | 适用于无 NVIDIA GPU 但需要进行基于 CPU 并行计算的平台 |
| GPU串行版 | 适用于大部分带有单、多个 NVIDIA GPU 的计算机或平台,无需安装MPI |
| GPU并行版 | 适用于配备多GPU的计算机或超算平台(平台需支持多GPU间通信) |
acpype
这是一个用于转换Amber生成的力场文件的Python脚本,在用于生成配体拓扑文件时使用,必须安装AmberTools方可使用
pip install acpypeVMD
一个经典的可视化软件,官方下载地址:
https://www.ks.uiuc.edu/Development/Download/download.cgi?PackageName=VMD
建议先别使用2.0,还在测试阶段
Xmgrace
官网地址 :Grace Home
或者使用包管理器安装(推荐,对于大多数人足够了)
sudo apt-get install grace
Xmgrace(也称为 Grace)是一款开源功能强大的交互式二维绘图工具,广泛应用于科学计算和数据分析领域,它支持多种操作系统,包括 Linux、Windows 和 macOS。
在Gromacs中许多分析工具的默认输出格式为.xvg,为xmgrace的默认格式,可用该软件打开或者通过脚本分析或转换。使用grace打开运行或者使用xmgrace打开图形界面
简单教程:GROMACS教程:Xmgrace学习笔记|Jerkwin
其他工具
Pymol
DuIvyTools:GROMACS模拟分析与可视化工具 (可选)
这是由佳佳辉大佬开发的一套python3的库--DuIvyTools,缩写DIT,用于便捷快速的可视化与Gromacs模拟工具。在本次教程中接下来使用这个工具进行部分可视化的操作,但并非唯一方法,绘图大佬选择自己喜欢的方法即可。注意,该工具包需要在可交互式界面下使用
如果您在研究中使用了 DuIvyTools,请通过 doi 引用:CharlesHahn/DuIvyTools: Confusion, that's my epitaph
安装方法:
pip install DuIvyTools运行中可能会遇到字体缺失问题,“findfont: Font family 'Arial' not found.”,安装即可 :
sudo apt-get install ttf-mscorefonts-installer中文教程地址:Welcome to DuIvyTools’s documentation! — DuIvyTools 0.6.0 documentation
分子动力学模拟
概念说明
周期性边界条件(Periodic Boundary Conditions,PBC):是一种重要的技术,用于模拟无限大的系统,从而避免因有限模拟盒子带来的边界效应 。
周期性边界条件的核心思想是将有限的模拟盒子在所有空间方向上无限复制,形成一个无限大的晶格。在这种情况下,当一个粒子从盒子的一边移出时,它会从另一边重新进入,从而保持粒子总数不变。这种设置使得粒子之间的相互作用类似于在无限大的系统中进行,从而能够更准确地模拟体相性质。
上面的说法是较为专业准确的,以下是我的一些个人理解,不一定完全正确,简单来说,分子模拟受限与算力与力场算法,无法做到与现实一致的模拟。但对于哪些具有规律性、周期性的物种(液体、气体、晶体等),我们可以通过规律将其划分为无数的“小盒子”,每一个小盒子都是类似的,是它们规律的“单元”,它们全部加一块就相当于原本的整体。
而每一个小盒子是一个有限空间内的模拟,但在模拟的过程中许多原子可能跑出我们的“小盒子”,此时如果直接将它们移出体系对于整个体系来说是不合理的,所以从整个体系来看,总的原子应当是不变的,所以设定当一个粒子从盒子的一边移出时,它会从另一边重新进入,从而保持粒子总数不变。(就类似与三体中描述的小宇宙,每一面都是与另一面相连接的)
拓扑化:通常指生成分子的拓扑文件(Topology File),这是模拟中一个非常重要的步骤。拓扑文件包含了分子系统的详细信息,包括原子类型、原子之间的键连接关系、力场参数等,这些信息是进行分子动力学模拟的基础
简单的说,就如同用乐高积木搭建一个复杂的模型。每个乐高积木块代表一个原子,而积木之间的连接方式(插槽和凸起)代表原子之间的化学键。在搭建乐高模型之前,你需要知道每个积木块的形状、大小,以及它们之间如何连接。
同样,在分子动力学模拟中,拓扑化的作用就是告诉计算机每个原子是什么类型(比如碳原子、氧原子),以及它们之间是如何连接的(比如通过单键、双键)。乐高积木的插槽和凸起有特定的规则,决定了哪些积木可以连接在一起。在分子动力学模拟中,拓扑文件就像这些规则,定义了原子之间的相互作用(比如键的强度、角度的大小)。而不同的力场对于同一个分子的拓扑化具有差异
文件说明
分子拓扑结构(Molecular Topology)是指分子中各个原子之间的连接关系和空间排列方式。它描述了分子的骨架结构,包括原子之间的键连接、键的类型、键角、二面角等信息,但不涉及具体的几何坐标(几何坐标通常由坐标文件如 .gro 或 .pdb 提供)
在 GROMACS 中,分子的拓扑结构文件(Topology File)是一个非常重要的输入文件,它定义了分子的化学结构和物理性质。拓扑文件通常以 .top 为扩展名,包含了以下关键信息:
1. 分子的组成
原子类型(Atom Types):定义了分子中每个原子的类型,例如碳原子、氧原子等。这些类型通常与力场参数相关联,用于计算非键相互作用。
原子名称(Atom Names):每个原子的唯一标识符,例如 C1、O2 等。
残基名称(Residue Names):定义了分子的残基结构,例如蛋白质中的氨基酸残基或核酸中的核苷酸残基。
分子类型(Molecule Types):定义了分子的类型,例如蛋白质、溶剂分子等。
2. 化学键
键(Bonds):定义了原子之间的化学键,包括键的类型(如单键、双键)和键的参数(如键长、键能)。
角(Angles):定义了三个原子之间的角,包括角的类型和参数(如键角、角能)。
二面角(Dihedrals):定义了四个原子之间的二面角,包括二面角的类型和参数(如二面角的势能函数)。
3. 非键相互作用
范德华力(Van der Waals Interactions):定义了原子之间的范德华力参数,用于计算非键相互作用。
电荷(Charges):每个原子的电荷值,用于计算库仑相互作用。
4. 其他信息
分子数量(Number of Molecules):定义了系统中每种分子的数量。
约束(Constraints):定义了需要约束的键或角,例如使用 LINCS 或 SHAKE 算法约束的键。
位置限制(Position Restraints):定义了某些原子的位置限制,通常用于模拟的初始阶段。
单独蛋白模拟
准备结构文件
这边笔记中文件的命名采用官方格式,实际使用中可以不同,但一定要有辨识性,后期文件数量将非常多。接下来以protein作为示例文件名称,使用请根据实际情况更改
首先由于下载的PDB结构大部分为结晶结构解析,我们首先需要去除水分子,可以使用可视化软件,或者使用文本编辑器一类的进行操作,Gromacs同样提供了一种便捷去除水分子的命令:
grep -v HOH protein.pdb > protein_clean.pdb注意,这种方法并不是普遍适用的(例如形成紧密结合或其他功能活性位点水分子,grep命令可参见此处)。
生成拓扑文件与力场选择
这一步主要是使用pdb2gmx工具将分子拓扑化
pdb2gmx的参数很多,但是常用的只有以下几个:
- -f 指定你的坐标文件,可以是pdb、gro、tpr等等包含有分子坐标的文件;
- -o 输出文件,也就是处理过的分子坐标文件,同样可以是pdb、gro、g96等文件类型;
- -p 输出拓扑文件,pdb2gmx读入力场文件,根据坐标文件建立分子系统的拓扑;
- -water 指定水模型(如
spc、spce、tip3p等)。默认为spce - -ignh 舍弃分子文件中的H原子,因为H原子命名规则多,有的力场不认;
- -his 独个指定HIS残基的质子化位置
gmx pdb2gmx -f protein_clean.pdb -o protein_processed.gro -water spce -ignh这里我们使用较为简单的参数, 还可以使用以下参数手动定义输出的top和itp文件名称
-p:指定输出的拓扑文件(.top格式),默认为topol.top-i:指定输出的包含分子拓扑的.itp文件,默认为posre.itp
输入以上命令后,会出现以下显示,提示你选择一个力场,力场的选择十分重要,要针对模拟的情况合理选择力场,这边我们使用6号力场
Select the Force Field:
From '/usr/local/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)这边完成拓扑化后,我们将得到以下三个格式的文件:
| 文件格式 | 用途 |
| .top | 分子的拓扑结构文件 |
| .itp | 位置限制文件 |
| .gro | 处理后的结构文件 |
这边简单介绍一下Gromacs原生支持的力场,不感兴趣的可以先跳过:
AMBER(Assisted Model Building and Energy Refinement)既是指一组用于模拟生物分子的分子机械 力场,也是一套分子模拟程序。
可以使用以下信息找到有关力场的信息:
- AMBER 力场- AMBER 力场的背景
- AMBER 程序- 有关 AMBER 分子模拟程序套件的信息
- ANTECAMBER/GAFF - 广义 Amber 力场 (GAFF),旨在提供适用于与 AMBER 蛋白质/核酸力场兼容的小分子的参数。它可以与 AMBER 一起使用,也可以通过 antechamber 软件包(也是单独发布的)获取。(这是接下来进行蛋白与配体时使用的方法)
CHARMM(Chemistry at HARvard Macromolecular Mechanics)是一套力场,同时也是一个用于分子动力学模拟和分析的软件包。它包含联合原子力场(CHARMM19)和所有原子力场(CHARMM22、CHARMM27、CHARMM36) 。CHARMM27 力场已移植到 GROMACS 并获得官方支持。CHARMM36 力场文件可从MacKerell 实验室网站获取,该网站定期更新 GROMACS 格式的 CHARMM 力场文件。
GROMOS是一个用于研究生物分子系统的通用分子动力学计算机模拟软件包。它还集成了涵盖蛋白质、核苷酸、糖等的自身力场,可应用于从玻璃、液晶到聚合物、晶体以及生物分子溶液等各种化学和物理系统。(Gromacs与Gromos之间的爱恨情仇:D,两者曾是因理念不同分出来的两个分支,而Gromacs一直计划移除Gromos力场)
溶剂化
分为两个步骤,首先定义一个盒子,然后将盒子填充溶剂
使用 editconf 模块定义盒子大小
gmx editconf -f protein_processed.gro -o protein_newbox.gro -c -d 1.0 -bt cubic-c 将蛋白放置与盒子中心
-d 1.0 使蛋白距离盒子边缘至少1.0 nm
-bt cubic 盒子类型定义为立方体
使用 solvate 模块在盒子中填充溶剂
gmx solvate -cp protein_newbox.gro -cs spc216.gro -o protein.gro -p topol.top-cp 指定溶质的结构文件,这里选择刚刚editconf生成的文件
-cs 指定溶剂的结构文件,spc216.gro是标准 GROMACS 安装的一部分,它是一个通用的平衡的三点溶剂模型,还可作为 SPC、SPC/E 或 TIP3P 的配置,因为它们都是三点水模型。(因为短暂的平衡过程即可消除模型之间的细微差异。其他溶剂以及混合溶剂也受支持,对溶剂类型的唯一限制是,一个溶剂分子只能包含一个残基)
-o 指定输出文件,这里为protein.gro
-p 指定拓扑文件的名称(topol.top),以便solvate进行修改
在完成后查看.top文件会发现在底部[molecules]中增加了SOL(溶剂)这一项。如下图所示
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10832 solvate 指令所做的就是记录它加入了多少水分子,然后把它写在你的拓扑结构上,以反映已经发生的变化。请注意,如果您使用任何其它(非水)溶剂,solvate 不会对您的拓扑结构做出这些更改(意味着你需要手动修改 .top 文件)。
添加离子(Adding Ions)
这一步的目的一方面是生物体系通常是电中性的,因此在模拟中也需要确保体系的电荷平衡。如果体系中的蛋白质或其他分子带有净电荷,就需要添加相应的抗衡离子来中和这些电荷;另一方面可以通过改变溶剂,模拟不同溶剂环境下的分子状态。
grompp需要附加的输入文件 .mdp (分子动力学参数文件)。grompp 将把 .mdp 文件中指定的参数与坐标和拓扑信息组合在一起,生成一个 .tpr 文件。在本例中,只进行简单的能力最小化操作,以下为本例mdp 文件,创建文件复制即可(对于大多数情况适用)。mdp文件中的参数是多样化的,要根据实际情况选择合适的参数。
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
energygrps = system ; Which energy group(s) to write to disk
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; long range electrostatic cut-off
rvdw = 1.0 ; long range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions首先使用grompp将mdp、gro、top 打包成Gromacs输入文件tpr
gmx grompp -f em.mdp -c protein_solv.gro -p topol.top -o ions.tpr现在可以将生成的tpr文件用于 genion 指令添加离子:
gmx genion -s ions.tpr -o protein_solv_ions.gro -p topol.top -pname NA -nname CL -neutral-s 输入结构/状态文件
-o 生成gro文件输出
-p 处理拓扑文件进行水分子的移除和离子的添加
-pname 定义正离子名称
-nname 定义负离子名称
-neutral 只添加必要的离子来中和蛋白质上的净电荷,通过添加正确数量的负离子。
用 -pname 和 -nname 指定的离子名称在以前的GROMACS版本中是与力场相关的,但在4.5版本中进行了标准化。指定的离子名称总是元素符号的全部大写字母(这也是[ moleculetype ]的名称),然后写入拓扑文件。根据力场的不同,残留物或原子名可能会附加或不附加电荷的符号(+/-)。不要在 genion 命令中使用 atom 或 residual 名称,否则您将在后续步骤中遇到错误。
能量最小化
溶剂化且电荷中性的系统现在已经组装好了,在我们开始动力学之前,我们必须确保系统没有空间冲突或不适当的几何形状。我们需要通过一种称为能量最小化(EM)的过程来放松结构。
组装tpr文件
同样使用的是上面的em.mdp
gmx grompp -f em.mdp -c protein_solv_ions.gro -p topol.top -o em.tpr在运行 genbox 和 genion 时前,确保你已经更新了你的 topol.top(指的是已经在文件中添加了相应的分子的数据),否则你会得到很多糟糕的错误消息(“坐标文件中的坐标数与拓扑文件不匹配”等)。
运行能量最小化
我们现在准备调用 mdrun 来执行EM:
gmx mdrun -v -deffnm em-v 将每一步都将进度输出到到屏幕上
-deffnm 定义输入和输出的文件名。如果与上一步打包时输出时定义的名称不一致,则必须使用 mdrun -s 来明确指定其名称。将得到以下文件:
| 文件格式 | 描述 |
| em.log | 文本日志文件 |
| em.edr | 二进制的能量文件 |
| em.trr | 二进制全精度轨迹文件 |
| em.gro | 能量最小化结构文件 |
分析能量最小化情况
em.edr 文件包含了 GROMACS 在EM期间收集的所有能量项。你可以使用 GROMACS 的 energy 指令分析任何 .edr 文件:
gmx energy -f em.edr -o potential.xvg使用xmgrace查看图像
xmgrace potential.xvg平衡 (Equilibration)
平衡常分两阶段进行。
NVT
第一阶段是在NVT系综下进行的(恒定的粒子数,体积和温度)。这个系综也称为“等温等容系综”或“正则系综”。这样一个过程的时间框架依赖于系统的内容,但在NVT中,系统的温度应该在期望的值上达到一个稳定的水平。如果温度还没有稳定下来,则需要额外的时间(来平衡温度)。通常,50-100 ps就足够了,本教程将进行100 ps的NVT平衡
title = OPLS Lysozyme NVT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is off
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed在运行中如果提示有警告,在确定无影响的情况可添加参数忽略(-maxwarn <警告数量>)
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
-f 指定模拟参数文件(.mdp),包含模拟的控制参数,如时间步长、截断半径等。
-c 指定输入的结构文件(如 .gro 或 .pdb),包含初始原子坐标
-p 指定拓扑文件(.top),包含分子类型、原子类型、力场参数等
-o 指定输出的二进制运行输入文件(.tpr),该文件是 gmx mdrun 的输入
-r 指定参考结构文件,用于位置约束或自由能计算
-maxwarn:设置允许的最大警告数量。默认值为 0,即不允许任何警告。
通过energy分析恒温过程:
gmx energy -f nvt.edr -o temperature.xvg在提示符下输入“16 0”,选择系统温度并退出
查看图像结果
xmgrace temperature.xvgNPT
前一步的NVT平衡稳定了系统的温度。接下来还须稳定系统的压力(以及密度)。压力平衡是在NPT系综下进行的,其中粒子数、压力和温度都是恒定的。该系综也称为“等温等压系综”,与实验条件最为相似。
NPT 平衡的.mdp文件可以在这里找到。它与用于NVT平衡的参数文件没有很大的不同。注意增加了压力耦合部分,使用的是Parrinello-Rahman压力耦合方法
title = OPLS Lysozyme NPT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = yes ; Restarting after NVT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = no ; Velocity generation is off 其他的一些变化:
- continuation = yes:继续从NVT平衡阶段进行模拟;
- gen_vel = no:速度是从轨迹中读取的(见下)。
注意,我们现在使用了 -t 以包括来自NVT平衡的检查点文件(这个文件包含继续模拟所需的所有状态变量)。为了保持NVT期间产生的速度,我们必须使用这个文件。坐标文件(-c)是NVT仿真的最终输出文件。
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx mdrun -deffnm npt
分析恒压过程
gmx energy -f npt.edr -o pressure.xvg
在提示符下输入“18 0”,并使用xmgrace查看结果
xmgrace pressure.xvg分析密度变化
gmx energy -f npt.edr -o density.xvg随后输入"24 0",并使用xmgrace查看结果
xmgrace density.xvg开始分子动力学模拟(Production MD)
在两个平衡阶段完成后,现在已经准备好解除位置限制,并运行MD进行数据收集。利用检查点文件来进行 grompp。我们将运行一个 1 ns MD 模拟,可以通过修改nsteps参数来调整总步数
title = OPLS Lysozyme NPT equilibration
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 500000 ; 2 * 500000 = 1000 ps (1 ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress bulky .trr file by specifying
nstvout = 0 ; 0 for output frequency of nstxout,
nstfout = 0 ; nstvout, and nstfout
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; save compressed coordinates every 10.0 ps
compressed-x-grps = System ; save the whole system
; Bond parameters
continuation = yes ; Restarting after NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; Velocity generation is off 将mdp、gro、cpt、top打包形成tpr文件
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
输入命令开始运行模拟
gmx mdrun -deffnm md_0_1 -v
- deffnm 定义输入和输出的文件名
- v 显示详细输出
完成模拟后可以先查看后续章节《模拟后处理与模拟轨迹可视化》的内容学习
蛋白与配体模拟
经过上面的学习,我们以及掌握了基础的单独蛋白的分子动力学模拟,解析我们需要学习如何模拟配体与蛋白共同模拟的情况。看上去很简单但实际面临的问题是,配体在 GROMACS 提供的任何力场中都不是可识别的实体,因此如果尝试将其传递给 pdb2gmx将会报错。只有当力场的 .rtp(残基拓扑)文件中存在构建块条目时才能自动组装拓扑。由于正常情况不太可能,我们将分两步准备系统拓扑:
- 使用 pdb2gmx 准备蛋白质拓扑
- 使用外部工具准备配体拓扑
准备配体文件
1.从复合体中分离蛋白与配体
由于我们将分别准备这两个拓扑结构,首先需要将蛋白和配体保存到单独的坐标文件中,这里有多种方法:
1.在开始前就使用Pymol等软件将两者分开保存
2.使用grep等文本工具将二者分开
例如一下示例,实际使用请根据情况修改选择:
grep "PROA" input.pdb > receptor.pdb
grep "LIG" input.pdb > ligand.pdb2.对于没有复合体的情况
如果没有复合体的情况,首先需要使用分子对接将配体与蛋白进行对接,得到合理的复合体文件,至于分子对接,目前已有大量成熟的软件,这边不额外进行介绍,如有需要可参考网上大量教程或本人另外一篇AutoDock4的笔记:AutoDock学习笔记--以win下AutoDock4为例_autodock运行显示python-CSDN博客
配体拓扑化
现在可以开始处理配体了,但对于力场无法自动识别的物质,该如何设计参数呢?正确处理配体是分子模拟中最具挑战性的任务之一。力场作者花费数年时间推导自洽的力场,而将新物质引入此框架并非易事。任何新物质的力场参数都必须以与原始力场一致的方式推导和验证。
Gromacs官方提供了一些参考的生成配体拓扑的工具,但由于时间久远,这边参考了另外一位大佬的文章,几种生成有机分子GROMACS拓扑文件的工具: 几种生成有机分子GROMACS拓扑文件的工具 - 思想家公社的门口:量子化学·分子模拟·二次元
| Sobtop | 由大佬们开发的主要产生GAFF/GAFF2、AMBER力场的拓扑文件,兼顾便利和灵活 |
| acpype | 需要安装AmberTools,较为臃肿,且只适合产生GAFF能描述的那些有机和极少数无机体系 |
| Automated Topology Builder | 它生成的原子电荷、确定的参数的可靠性也只是一般,不能保证总是很理想。 提交到ATB之前建议先搜索一下分子库,如果已经有的话就直接用 |
| LigParGen | |
| MKTOP | |
| OBGMX | |
| SwissParam | |
| ztop | |
| CGenFF | 能够生成CGenFF力场的GROMACS拓扑文件的在线工具,学术用户必须用edu邮箱才能注册 |
以下表格是Gromacs官方对于不同物质分子的建议力场:
| Molecule/Ion Type | Force Field |
|---|---|
| protein | ff19SB |
| DNA | OL21 |
| RNA | OL3 |
| carbohydrates | GLYCAM_06j |
| lipids | lipids21 |
| organic molecules (usually ligands) | gaff2 |
| ions | •should be matched to water model; see force fields for ions for further discussion |
| water model | •should be matched to atomic ions; common water models include tip3p, spc/e, tip4pew, and OPC |
添加氢原子
首先需要给配体加上氢,可以使用ruduce或obabel工具
reduce更适合处理生物大分子(如蛋白质、核酸),特别是在需要考虑生物化学环境(如 pH 值)时,但质子化状态调整依赖于预设的规则和参数,这些规则可能不适用于所有生物化学环境。obabel更适合处理通用化学分子,支持多种格式和操作,适合需要灵活处理分子数据的场景。会根据 pH 值调整分子的质子化状态,但这种调整是基于原子级别的,而不是分子整体的且处理生物大分子时可能不如reduce精确
reduce input.pdb > input_h.pdb -H常用参数:
-H:添加氢原子-build:根据残基名称和编号添加氢原子-n:不添加氢原子,仅优化现有氢原子的位置-pH:指定 pH 值以调整质子化状态(默认为 7.0)-keep:保留输入文件中的氢原子-output:指定输出文件名-dir:指定输出文件的目录
或者使用obabel,注意-O是大写
obabel input.pdb -O input_h.pdb -h常用参数:
-i<format>:指定输入文件的格式-o<format>:指定输出文件的格式-O<filename>:指定输出文件名-h:添加氢原子-p:指定pH 值添加氢原子--addH:添加氢原子--gen3D:生成 3D 坐标--gen2D:生成 2D 坐标--addNonPolarH:仅向非极性原子添加氢原子--addPolarH:仅向极性原子添加氢原子-f:从指定的分子编号开始处理-l:在指定的分子编号处结束处理-m:生成多个输出文件,适用于批量转换
拓扑化
使用AmberTools的antechamber模块将配体拓扑化
antechamber -i ligand_new.pdb -fi pdb -o ligand.mol2 -fo mol2 -c bcc -rn ligand
输入输出相关参数
-i:指定输入文件的名称。-fi:指定输入文件的格式,如mol2、pdb、gout等。-o:指定输出文件的名称。-fo:指定输出文件的格式,如mol2、prepi、frcmod等。
电荷相关参数
-c:指定电荷计算方法,如bcc(基于 AM1 的电荷计算)或resp(基于高斯计算的静电势拟合电荷)。-nc:指定分子的净电荷。如果分子本身带电,需要手动设置该参数。-gk:在使用高斯计算静电势时,可以指定高斯的关键词,如#HF/6-31G* SCF=tight Test Pop=MK等。
其他参数
-pf:设置为y可以启用多核计算,加快处理速度。-rn:为生成的分子指定一个名称,方便在后续处理中识别。-gm:在使用高斯计算时,可以指定高斯的内存分配,如%mem=4096MB。-gn:在使用高斯计算时,可以指定高斯的核数,如%nproc=8
格式转换
由于使用antechamber工具生成的立场文件无法直接用于Gromacs,我们需要使用acpype这个Python脚本将其转换为Gromacs兼容的力场文件
acpype -i PNPC.mol2输入输出相关参数
-i:指定输入文件名,支持.pdb、.mol2或.mdl格式。-p:指定 Amber 的.prmtop文件名,通常与-x参数一起使用。-x:指定 Amber 的.inpcrd文件名,通常与-p参数一起使用。-b:指定项目的基名(basename),用于生成输出文件夹和文件名。-o:指定输出拓扑文件的格式,可选值为all(默认)、gmx、cns或charmm。
力场和原子类型相关参数
-a:指定原子类型,可选值为gaff(默认)、gaff2、amber(AMBER14SB)或amber2(AMBER14SB + GAFF2)。-q:指定 AM1-BCC 电荷计算程序,可选值为sqm(默认)、divcon或mopac。-k:指定 MOPAC 或 SQM 的关键词,需要放在引号内。
电荷相关参数
-c:指定电荷计算方法,可选值为gas、bcc(默认)或user(用户在.mol2文件中提供的电荷)。-n:指定分子的净电荷(整数),默认为 0。-m:指定多重性(2S+1),默认为 1。
其他参数
-f:强制重新计算拓扑文件,即使已存在。-d:调试模式,保留所有临时文件。-z:为 GROMACS 4.0 写入 RB 二面角参数。-t:为 CNS/XPLOR 写入类似 allhdg 的参数(实验性)。-e:指定使用的引擎,可选值为tleap(默认)或sleap。-s:设置 SQM/MOPAC 的最大运行时间(秒),默认为 10 小时。
在完成后将得到一个文件夹,我们接下来需要的文件如下,分别是GMX兼容的配体的.itp和.gro,可以将这两个文件单独复制到工作目录下
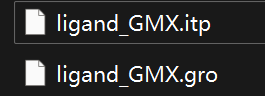
组装蛋白与配体
经过前面的步骤得到了蛋白和配体的各自拓扑文件,现在需要我们手动将其组装成复合物
(建议)复制一份之前生成的蛋白质的gro文件,并将其改名(例如complex)
组装gro文件
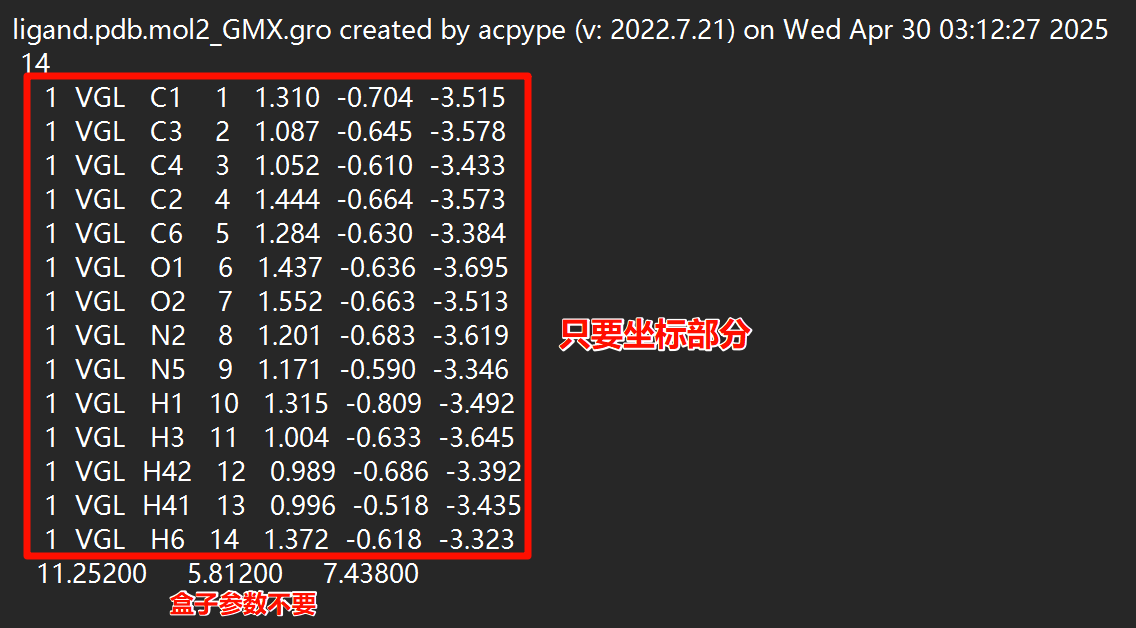
使用文本编辑器一类打开ligand_GMX.gro文件,将其原子坐标部分全部复制,并黏贴至complex.gro的原子坐标部分最后,注意蛋白原本的盒子信息要在最后一行
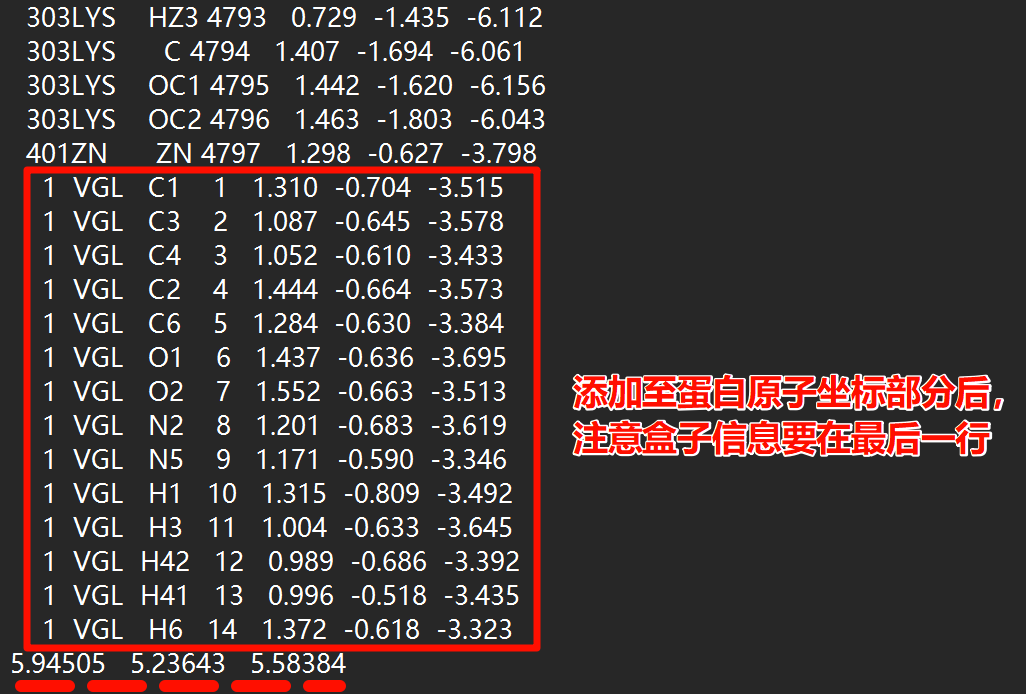
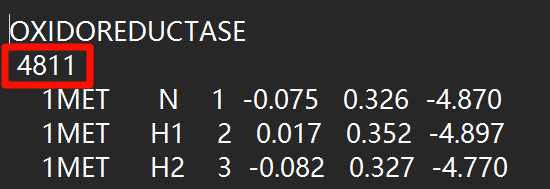
并且由于添加了原子,需要将gro文件第二行的数量修改为增加后的原子总数
修改top文件
组装完gro文件后,我们还需要修改top文件,告诉它我们增加的配体的分拓扑文件(itp)是什么,在top文件中添加以下定义(建议添加在力场定义下,如下图所示,错误的位置可能导致后面奇怪的报错!!本人遇到过,具体原因不太了解,可能与其严格的文件要求有关)
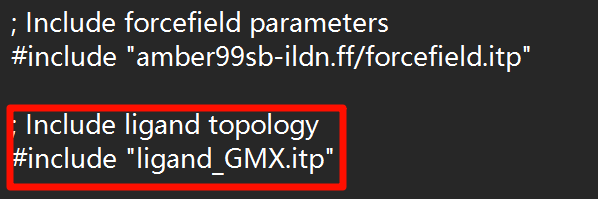
使用时注意将"ligand_GMX.itp"修改为实际文件名称
; Include ligand topology
#include "ligand_GMX.itp"并在top文件的末尾修改[ molecules ]部分,加入我们添加的分子名称,例如下图,添加了ligand。注意!这里的分子名称是之前配体拓扑化时定义的,如果不确定可查看“ligand_GMX.itp”中的[ moleculetype ]来确定。
[ molecules ]
; Compound #mols
Protein_chain_A 1
ligand 1溶剂化 、能量最小化
这部分操作与之前单独蛋白一致,创建盒子,随后填充溶剂,添加离子平衡电荷,以及能量最小化,这里不再赘述。
NVT、NPT平衡
平衡蛋白质配体复合物与平衡任何其他蛋白质水溶液体系一样. 但需要有些特殊考虑:
- 对配体施加限制
- 处理温度耦合组
对配体施加限制
gmx genrestr -f PNPC_GMX.gro -o posre_PNPC.itp -fc 1000 1000 1000-o:指定输出的.itp文件,该文件包含位置约束或距离约束的定义-
-fc:指定力常数(单位为 kJ/mol nm²),可以分别设置 x、y、z 方向的力常数,也可以使用一个各向同性的值。这边我们使用1000 1000 1000,这个参数较为常用
在top文件中再添加一个定义(最好就添加在之前添加的配体拓扑文件定义下,原因如上),注意将“posre_ligand.itp”修改为上一步生成的实际文件名称:
; Ligand position restraints
#ifdef POSRES
#include "posre_ligand.itp"
#endif处理温度耦合组
对只有几个原子的组在控制其动能的涨落时, 温度耦合算法不够稳定,不要对体系中的每一单个物种使用独立的耦合。创建一个特殊的组, 其中包含蛋白质和配体。
gmx make_ndx -f em.gro -o index.ndx通过下面的命令合并"Protein"和"ligand",其中">"是make_ndx的提示符;注意!请根据实际显示选择,不同情况显示的对象和数字可能不同
> 1 | 13
> q准备好以上步骤后,即可开始NVT、NPT平衡,与之前单独蛋白操作一致
运行模拟
这一步与前面一致
模拟后处理与模拟轨迹可视化(待更新)
对于模拟生成的文件,有多种方法进行可视化,本次采用VDM进行简单的可视化操作
周期性边界条件的处理
这一步也称为恢复周期性边界,使用GROMACS完成模拟后, 有时需要对轨迹的周期性边界条件(PBC)进行处理. 可能的目的主要有两个:
- 为了观看轨迹时分子保持完整, 不产生断键或过长的键, 同时未解离的复合物保持在一起
- 为了对选定的中心分子进行居中, 以便进行后面的分析
二聚体及多聚体等特殊的分子处理较为麻烦且不同,这边主要以单体为例进行周期性边界条件的处理
模拟完成得到轨迹后, 按照经典的轨迹处理步骤, 先保证体系中每个分子完整:
gmx trjconv -s npt.tpr -f prod.xtc -o prod_whole.xtc -pbc whole
-s 指定结构文件,用于提供拓扑信息和初始构型。
-f 指定的轨迹文件
-o 输出文件周期性边界条件处理选项,whole:仅修复断裂的分子
-pcb whole
这边如果只是想快速做图,也可以使用VMD中的指令调整显示
pbc wrap -compound res -all
pbc box #显示盒子加载分子文件(.gro)
启动VMD软件,在初始界面点击File>New Molecule打开面板选择Browse加载模拟结果的.gro文件,点击Load加载至VMD中(对于多数情况Determine file type选择为Automatically就可以了,默认选择)
删除第一帧重复帧
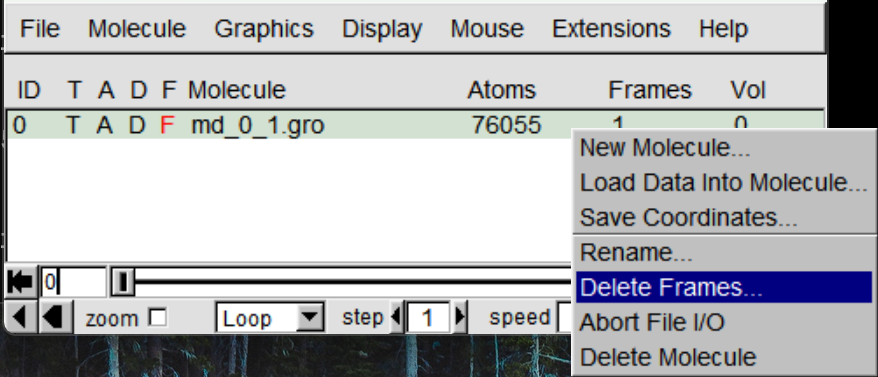
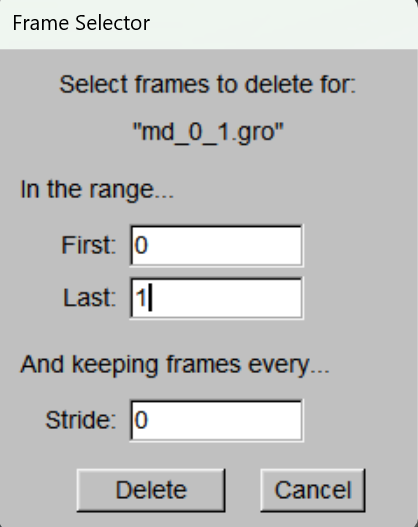
加载完成文件后,主窗口将显示该文件信息,左键选择对应分子随后右键,在出现的菜单中点击Delete Frames,在出现的面板中修改Last栏为1,点击Delete删除第一帧
加载轨迹文件(.xtc)
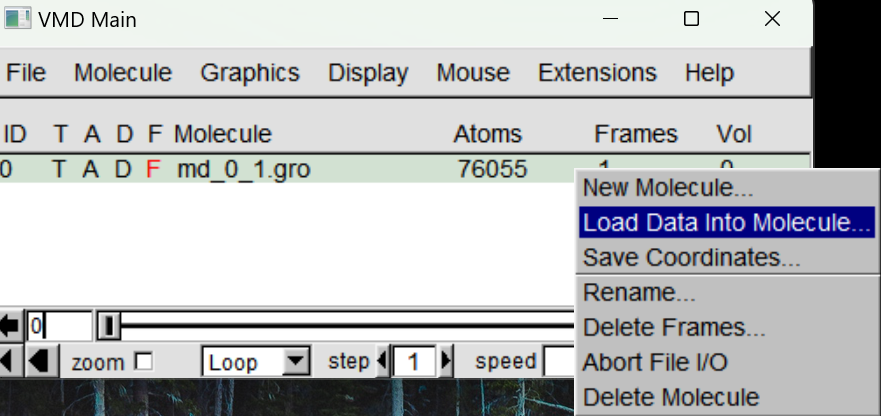
同样左键选中后右键选择Load Data into molecule选项,通过Browse加载运行生成的.xtc文件,点击Load加载值VMD中(这里可以选择Load all at once选项,加载的更快)
修改优化显示方式
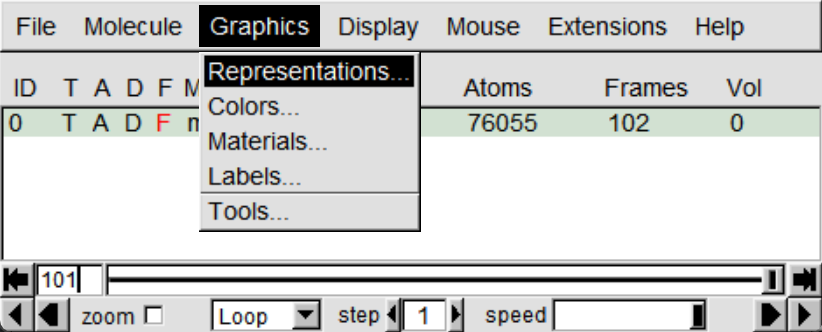
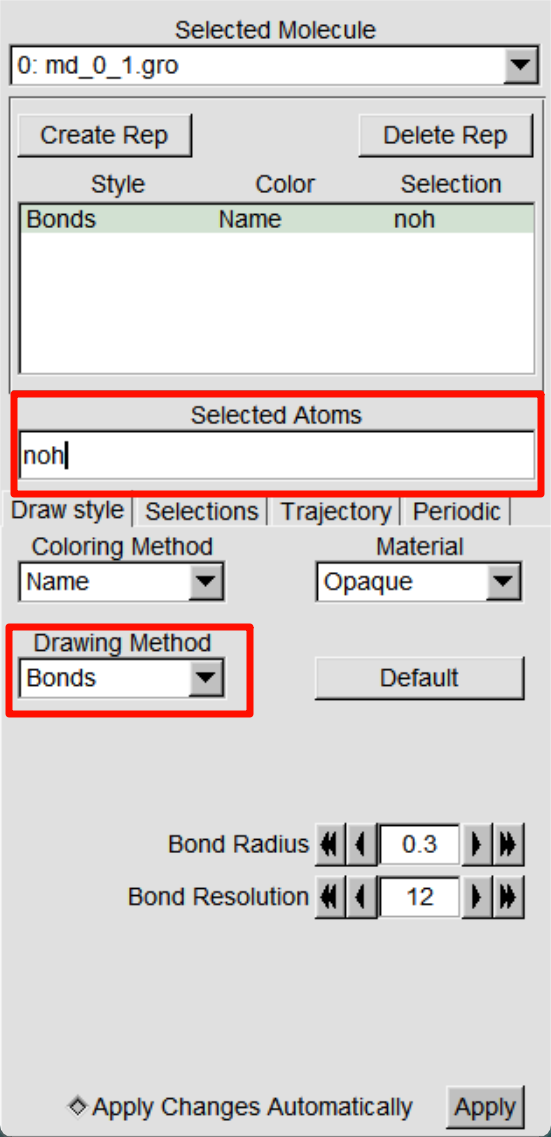
完成以上操作后,成功将轨迹在VMD中进行了可视化,但会发现溶剂分子挡住了蛋白,我们可以点击Graphics >Representations打开面板,在Selected Atoms栏中修改为noh,点击应用修改显示原子,还可以修改Drawing Method选择合适的显示方式。
完成以上操作后,就可以再VMD的主窗口中通过鼠标操作(左键旋转、中键平移、滚轮缩放)好好欣赏你的分子动力学模拟结果了 。以及可以通过底部的操作栏来修改一些播放参数
![]()
附录
Gromacs中常用文件格式及其用途总览
| 文件格式 | 名称 | 用途 |
| .gro | GROMACS 坐标文件 (分子坐标文件) | 主要用于存储分子系统的坐标信息(以及可选的速度信息和盒子尺寸信息) |
| .top | 分子拓扑文件 | 包含的力场,键相互作用参数,非键相互作用参数,限制性参数,定义一些名称等 |
| .itp | 分拓扑文件 | 含某个特定分子的类型,不引用其他力场文件,同时包含[system],[molecule]等拓扑字节 |
| .rtp | 残基拓扑文件 | 该文件包含常见残基的力场信息,包括残基所含原子,成键种类等。使用pdb2gmx处理PDB文件时,程序按照PDB文件信息,在RTP文件中寻找对应的残基力场信息 |
| .ndx | 原子索引文件 | 该文件含原子的序号,当使用make_ndx程序生成索引文件时,可以定义不同的原子组,每组名下即是该组所含各个原子的序号 |
| .tpr | 运行输入文件 | 内容包含模拟的初始结构,分子拓扑,和所有的模拟参数。利用Grompp程序连接分子结构文件(.gro)、拓扑文件(.top)、分子动力学参数文件(.mdp)和索引文件(.ndx)(可选),即生成该文件。这个文件包含了开始使用 Gromacs 进行模拟的所有信息 |
| .trr | 二进制轨道文件 | mdrun 的输出文件是轨道文件和一个日志文件(.log)。它包含所有的坐标、速度、力和能量信息,就如在(.mdp)文件中指出的一样 |
| .log | 日志文件 | 使用mdrun运行时生成的日志文件 |
| .xtc | 单精度轨迹文件 | 文件较TRR和TRJ小,为常用分析文件。包含模拟系统中原子坐标,模拟时间,和模拟盒子信息 |
| .edr | 系统能量文件 | 该文件记录模拟输入文件中定义的能量组的各种相互作用能量等 |
| .xvg | 二维图标文件 | 二维绘图工具 xmgrace 的默认文件格式。它可以直接被 xmgrace 打开并用于生成图表 |
| .cpt | 模拟断点文件 | 模拟过程固定时间间隔产生,保存模拟系统所有信息。可以使用该文件重新在断点处开始模拟,也可以依靠该断点文件开始,并延长模拟计算 |
Gromacs常用命令
这边仅分析常用指令及其常用参数,详细内容请参考官方命令行手册:Command-line reference - GROMACS 2025.1 documentation
gmx pdb2gmx
gmx pdb2gmx -f(输入pdb文件)-o(输出gro文件)-p(输出top文件)-i(输出itp文件)-n(输出ndx)-ignh(舍弃H,系统自动加上)
输入输出文件相关参数
-f:指定输入的结构文件(如.pdb或.gro格式)。-o:指定输出的 GROMACS 格式坐标文件(如.gro或.pdb格式)。-p:指定输出的拓扑文件(.top格式)。-i:指定输出的包含分子拓扑的.itp文件。-n:指定输出的索引文件(.ndx格式)。-q:指定输出的包含电荷信息的文件(可选)。
力场和水模型相关参数
-ff:指定使用的力场名称(如charmm27、amber99等)。默认情况下,力场选择是交互式的,但可以通过此参数直接指定。-water:指定水模型(如spc、spce、tip3p等)。默认为spce。
氢原子处理相关参数
-ignh:忽略输入文件中的氢原子,程序会自动添加氢原子。-una:不添加任何氢原子。-heavyh:为重原子添加氢原子。-deuterate:将所有氢原子替换为氘原子。
残基处理相关参数
-ter:允许用户选择 N 端和 C 端残基的质子化状态。-lys:允许用户选择赖氨酸(LYS)的质子化状态。-arg:允许用户选择精氨酸(ARG)的质子化状态。-asp:允许用户选择天冬氨酸(ASP)的质子化状态。-glu:允许用户选择谷氨酸(GLU)的质子化状态。-gln:允许用户选择谷氨酰胺(GLN)的质子化状态。-his:允许用户选择组氨酸(HIS)的质子化状态。
其他参数
-chainsep:指定多链分子的链分离方式。-merge:指定是否合并多链分子。-angle:设置氢键角度的阈值(用于组氨酸的质子化状态判断)。-dist:设置氢键距离的阈值(用于组氨酸的质子化状态判断)。-posrefc:设置位置约束的力常数。-vsite:指定虚拟位点的类型。-renum:重新编号残基。-rtpres:保留残基类型文件中的定义
pdb2gmx更多参数:
gmx pdb2gmx - GROMACS 2025.1 documentation
gmx editconf
gmx editconf -f(输入gro文件)-bt(盒子类型,默认立方)-d(溶质分子与盒子之间的距离)-c(分子居中)
gmx editconf将通用结构格式转换为.gro或.pdb.g96
输入输出文件相关参数
-f:指定输入的结构文件(如.gro、.g96、.pdb等格式)。-n:指定索引文件(.ndx),用于选择特定的原子组。-o:指定输出的结构文件(如.gro、.g96、.pdb等格式)。-mead:生成用于 MEAD 电静力程序的特殊.pqr文件。
盒子操作相关参数
-bt:指定盒子类型,可选值包括:triclinic:三斜盒子。cubic:立方盒子。dodecahedron:菱形十二面体盒子。octahedron:截断八面体盒子。-box:设置盒子的向量长度(a, b, c)。对于立方、菱形十二面体或截断八面体盒子,仅需一个值。-angles:设置盒子向量之间的夹角(bc, ac, ab),仅与-box和三斜盒子相关。-d:设置溶质与盒子边界的距离。对于三斜盒子,使用系统在 x、y 和 z 方向的尺寸;对于其他盒子类型,尺寸设置为系统直径加上两倍指定距离。-c:将分子居中于盒子内(默认由-box和-d暗示)。-center:将系统的几何中心移动到指定的坐标 (x, y, z)。-pbc:移除周期性边界条件(粗略处理),使分子完整。
分子操作相关参数
-translate:将坐标平移指定的向量。-rotate:围绕 X、Y 和 Z 轴旋转指定的角度(单位为度)。-princ:将分子的主轴对齐到坐标轴,最长轴对齐到 x 轴。-scale:对盒子和坐标进行缩放。-density:通过缩放达到指定的密度(单位为 g/L)。-align:将指定组的主轴对齐到目标向量,可选中心点由-aligncenter指定。
其他功能参数
-resnr:从指定的编号开始重新编号残基。-label:为.pdb文件添加链标识符。-conect:在输出的.pdb文件中添加连接记录。
gmx editconf - GROMACS 2025.1 documentation
gmx solvate
gmx solvate -cp(输入gro文件,分子坐标)-cs(输入gro文件,盒子坐标)-o(输出gro文件)-p(输出top文件)
输入输出文件相关参数
-cp:指定溶质的结构文件(如.gro、.g96、.pdb等),通常是蛋白质或其他分子。-cs:指定溶剂的结构文件(如.gro、.g96、.pdb等),默认为 SPC 水模型(spc216.gro)。-p:指定拓扑文件(.top),用于更新溶剂分子的数量。-o:指定输出的溶剂化后的结构文件(如.gro、.g96、.pdb等)。
盒子设置相关参数
-box:指定盒子的尺寸(单位为纳米),格式为<x> <y> <z>。如果未指定,将使用溶质文件中的盒子尺寸。-shell:在溶质周围添加一层指定厚度(单位为纳米)的溶剂层。
溶剂化控制参数
-radius:默认的范德华半径(单位为纳米),默认值为 0.105。-scale:缩放范德华半径的因子,用于调整溶剂与溶质之间的距离,通常为 0.57。-maxsol:限制盒子中添加的最大溶剂分子数。如果设置为 0(默认值),则不限制。
其他参数
-[no]vel:是否保留输入文件中的速度信息。默认为不保留
gmx grompp
gmx grompp -f(输入mdp文件)-c(输入gro文件)-r(输入限制gro文件,这项要看自己使用的软件版本,旧版不强制,一般与-c传递gro文件同,复杂情况另说)-p(输入top文件)-o(打包输出tpr)
基本参数
-f:指定模拟参数文件(.mdp),包含模拟的控制参数,如时间步长、截断半径等。-c:指定输入的结构文件(如.gro或.pdb),包含初始原子坐标。-p:指定拓扑文件(.top),包含分子类型、原子类型、力场参数等。-o:指定输出的二进制运行输入文件(.tpr),该文件是gmx mdrun的输入。
高级参数
-r:指定参考结构文件,用于位置约束或自由能计算。-rb:指定 B 拓扑的参考坐标文件,用于自由能计算。-n:指定索引文件(.ndx),用于选择特定的原子组。-maxwarn:设置允许的最大警告数量。默认值为 0,即不允许任何警告。-pp:输出预处理后的拓扑文件,用于调试。-sort:对原子进行排序,以优化性能
gmx genion
是 GROMACS 中用于在模拟体系中添加单原子离子的工具。它通过随机替换溶剂分子来添加离子,确保体系的电中性或达到指定的离子浓度
gmx genion -s(输入tpr文件)-o(输出gro文件) -p(top文件) -pname(阳离子名称)-nname(阴离子名称)-neutral(自动中和所带电荷)
基本参数
-s:指定输入的.tpr文件,该文件由gmx grompp生成,包含体系的初始状态。-o:指定输出的结构文件(如.gro格式),包含添加离子后的体系。-p:指定拓扑文件(.top),用于更新离子的数量和类型。-n:指定索引文件(.ndx),用于选择特定的原子组。
离子相关参数
-pname:指定正离子的名称(如NA表示钠离子),默认为NA。-np:指定添加的正离子数量。-nname:指定负离子的名称(如CL表示氯离子),默认为CL。-
-nn:指定添加的负离子数量。 -neutral:自动添加离子以中和体系的净电荷。-conc:指定离子的浓度(单位为 mol/L),用于添加特定浓度的离子。
其他参数
-rmin:指定离子之间最小距离的阈值。-seed:设置随机数种子,用于控制离子的随机分布
gmx mdrun
gmx mdrun是 GROMACS 中的主要计算化学引擎。它不仅可以进行分子动力学模拟,还可以进行随机动力学、能量最小化、测试粒子插入或能量(重新)计算
gmx mdrun -deffnm “设置的输出文件名称”
如果模拟中途停止(并不是崩溃,没有任何报错的那种停止(比如你的terminal突然shutdown了或者断电了)),可以使用前一部分模拟产生的日志文件续跑(其中-cpi表示使用检查点文件,-append默认为 yes,表示将模拟结果续写到原文件中):
gmx mdrun -s “文件名”.tpr -cpi “文件名”.cpt -append
如果模拟正常结束后希望延长模拟时间,并输出到新的一系列名为md_0_2的文件中,可以:
gmx convert-tpr -s md_0_1.tpr -extend 1000 -o md_0_2.tpr
gmx mdrun -v -deffnm md_0_2 -cpi md_0_1.cpt -noappend
输入输出相关参数
-s:指定输入的.tpr文件,包含模拟的所有设置。-o:指定轨迹文件的输出名称(如.trr格式)。-x:指定压缩轨迹文件的输出名称(如.xtc格式)。-c:指定模拟结束时的坐标文件输出名称(如.gro或.pdb格式)。-e:指定能量文件的输出名称(.edr格式)。-g:指定日志文件的输出名称。-cpo:指定检查点文件的输出名称。-deffnm:设置输出文件的默认名字前缀,方便统一管理输出文件。-cpi:指定检查点文件(.cpt),用于从中断处恢复模拟。
并行计算相关参数
-nt:指定运行模拟的线程总数。-ntmpi:设置使用的 MPI 线程数。-ntomp:设置每个 MPI 线程使用的 OpenMP 线程数。-pin:控制线程/核心的固定模式,可选值包括on、off和auto。-gpu_id:指定 GPU 的 ID,用于 GPU 加速计算。-gputasks:分配 GPU 任务,指定每个 GPU 上的任务分配。
模拟控制参数
-nsteps:覆盖.mdp文件中的模拟步数设置。-maxh:指定最大运行小时数,超过该时间模拟会自动停止。-cpt:设置检查点文件的写入间隔(单位为小时)。-append:在继续模拟时,将输出追加到现有文件中。-noconfout:禁止在模拟结束时输出最终的坐标文件。
其他功能参数
-v:冗余模式,显示更多运行信息。-pforce:当原子受力超过指定值时,打印坐标和受力信息。-replex:设置复制交换模拟的交换频率。-reseed:重新设置随机数种子。- 并行化策略相关参数
-dd:设置区域分解(Domain Decomposition)的网格向量。-rdd:设置区域分解的最大距离。-rcon:设置约束连接的最大距离。-dds:设置动态负载平衡的单元格缩放因子。-npme:设置用于 PME 计算的线程数。
https://manual.gromacs.org/current/onlinehelp/gmx-mdrun.html
gmx trjconv
gmx trjconv -s(输入tpr文件)-f(输入xtc文件)-o(输出xtc文件)-pbc(PBC周期性边界) -centre(中心)
输入输出相关参数
-f:指定输入轨迹文件,支持.xtc、.trr、.gro、.g96、.pdb和.tng等格式。-s:指定结构文件(如.tpr、.gro、.g96、.pdb等),用于提供拓扑信息和初始构型。-n:指定索引文件(.ndx),用于选择特定的原子组。-o:指定输出文件名,支持多种格式,与输入文件格式类似。
时间相关参数
-b:指定从轨迹中读取的第一帧时间(单位为 ps,默认为 0)。-e:指定从轨迹中读取的最后一帧时间(单位为 ps,默认为 0)。-dt:指定输出帧的时间间隔(单位为 ps),用于减少输出帧数。-t0:设置输出轨迹的起始时间(单位为 ps)。-timestep:修改输入帧之间的时间步长(单位为 ps)。-dump:导出指定时间附近的帧。
轨迹处理相关参数
-
-pbc:处理周期性边界条件(PBC),可选值包括:
none:不处理 PBC。
mol:将分子的质心保留在盒子内。
res:将残基的质心保留在盒子内。
atom:将所有原子保留在盒子内。
nojump:防止原子在盒子中跳跃。
cluster:将所有原子聚集到质心附近。
whole:仅修复断裂的分子。-
-ur:设置单元格表示方式,可选值为rect(矩形)、tric(三斜晶胞)和compact(紧凑模式)。 -center:将原子质心居中于盒子内。-fit:将分子拟合到参考结构,可选值包括none、rot+trans(旋转和平移)、rotxy+transxy(仅在 xy 平面上旋转和平移)等。-skip:仅输出每第 n 帧。-sep:将每一帧输出为单独的.gro、.g96或.pdb文件。
其他参数
-box:设置新盒子的大小。-trans:将所有坐标平移指定的向量。-shift:将每一帧的坐标按帧号乘以指定向量进行平移。-ndec:设置输出.xtc文件的小数位数。-vel:读取和写入速度(如果可能)。-force:读取和写入力(如果可能)。-split:按指定时间间隔分割输出文件。-conect:在输出文件中包含连接信息。
nojump检查原子是否跳过盒子,然后将其放回原位。这样做的结果是所有分子都将保持完整(前提是它们在初始构象下是完整的)。注意,这确保了轨迹的连续性,但分子可能会扩散出盒子。此过程的起始构型取自结构文件(如果提供了文件),否则取自第一帧。cluster将选定索引中的所有原子聚类,使它们最接近簇的质心,并迭代更新簇的质心。请注意,只有当您确实拥有簇时,此方法才会给出有意义的结果。幸运的是,之后可以使用轨迹查看器进行检查。另请注意,如果您的分子断裂,此方法也将不起作用。
gmx energy
是 GROMACS 中用于从能量文件(.edr)中提取和分析能量成分的工具。
输入输出文件参数
-f:指定输入的能量文件(.edr)。-f2:指定第二个能量文件(可选),用于比较或组合数据。-s:指定输入的.tpr文件,用于获取额外的系统信息(可选)。-o:指定输出文件(.xvg格式),用于保存提取的能量数据。-viol:输出违反距离约束的统计信息(.xvg格式)。-pairs:输出选定原子对之间的距离统计信息(.xvg格式)。-corr:输出能量项的相关函数(.xvg格式)。-vis:输出能量项的可视化数据(.xvg格式)。-evisco:输出能量项的粘度相关数据(.xvg格式)。-odh:提取并绘制自由能数据(如 Hamiltonian 差异或导数)。
时间范围和精度控制
-b和-e:分别指定分析的起始时间和结束时间。-xvg:设置输出文件的格式,可选值包括xmgrace、xmgr、none等。-[no]w:是否覆盖已存在的输出文件。-[no]dp:是否使用双精度进行计算。
数据处理和统计分析
-[no]sum:是否输出能量项的总和。-nbmin和-nbmax:设置用于误差估计的块大小范围。-fluct_props:计算与能量波动相关的性质,如热容 Cp、Cv,热膨胀系数等。-nmol:设置体系中的分子数量,用于计算热容等性质。-feetemp:设置自由能计算的温度。-zero:将能量项的初始值设置为零。
其他功能
-fitfn:指定拟合函数类型,用于分析能量数据。-beginfit和-endfit:设置拟合的起始和结束时间。-acflen:设置自相关函数的长度。-[no]normalize:是否对输出数据进行归一化。
gmx rms
gmx rms -tu(时间单位)-s(tpr文件输入)-f(xtc输入)-o(输出xvg文件)
输入输出文件参数
-s:指定参考结构文件(如.tpr或.gro),用于比较。-f:指定轨迹文件(如.xtc或.trr),包含需要比较的结构。-f2:指定第二个轨迹文件,用于生成两个轨迹之间的比较矩阵。-n:指定索引文件(.ndx),用于选择特定的原子组。-o:指定输出文件(如.xvg),记录 RMSD 值。-mir:输出与参考结构镜像的 RMSD 比较结果。-m:生成一个.xpm格式的矩阵文件,包含轨迹中所有结构的比较值。-bin:对比较矩阵进行二进制转储。-bm:生成平均键角偏差的矩阵。
分析选项
-what:选择计算的参数类型,可选值包括rmsd(均方根偏差)、rho(尺寸无关的相似性参数)或rhosc(标度相似性参数)。-fit:控制结构之间的最小二乘拟合方式,可选值包括rot+trans(旋转和平移)、translation(仅平移)或none(不拟合)。-mw:是否使用质量加权,默认为开启。-pbc:是否考虑周期性边界条件,默认为开启。-prev:与前面指定帧数的结构进行比较。-skip:每隔若干帧计算一次 RMSD。
时间控制参数
-b:指定从轨迹文件中读取的第一帧时间。-e:指定从轨迹文件中读取的最后一帧时间。-dt:指定时间间隔,仅分析满足条件的帧。-tu:指定时间单位,可选值包括fs、ps、ns、us、ms、s。
其他参数
-max和-min:设置比较矩阵的最大和最小水平。-bmax和-bmin:设置键角矩阵的最大和最小水平。-nlevels:设置矩阵的水平数。-ng:指定计算 RMSD 的组数。
gmx make_ndx
gmx make_ndx -f(输入gro文件)-o(输出ndx文件)
gmx make_ndx 是 GROMACS 中用于创建和编辑索引文件的工具,索引文件(.ndx)用于定义特定的原子组,以便在模拟和分析中使用。以下是其常用参数和功能的总结:
基本参数
-f:指定输入的结构文件(如.gro、.g96、.pdb等),用于生成索引文件。-n:指定现有的索引文件(.ndx),可以在此基础上进行编辑。-o:指定输出的索引文件名称。-natoms:手动设置原子数量(默认从输入文件中读取)。-twin:复制所有索引组,偏移量为-natoms,适用于双层膜系统。
交互式命令
运行 gmx make_ndx 后,会进入交互式界面,用户可以通过以下命令操作:
r:按残基编号或名称选择残基。a:按原子编号或名称选择原子。chain:按链标识符选择原子。&和|:分别表示逻辑“与”和“或”操作。!:表示逻辑“非”操作。name:重命名索引组。del:删除指定的索引组。q:保存并退出
参考资料
https://zhuanlan.zhihu.com/p/25973205596
https://zhuanlan.zhihu.com/p/666826404
手把手教你用linux安装Gromacs(2024 GPU-CUDA)_gromacs安装-CSDN博客
https://zhuanlan.zhihu.com/p/650111719
GROMACS Tutorial 1-Lysozyme in Water-translated with notes-CSDN博客
https://zhuanlan.zhihu.com/p/479014455

















 3036
3036

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









