






1.LRT(likelihood ratio test)
1.1 原理介绍
似然比检验是用两个不同模型的似然函数作比,也就是似然比来检测某个假设是否有效的一种检验方法。一遍情况下,想要检测某个附加的限制函数是否正确(如限制了吸烟这个变量),用附加限制条件参数的模型,来构建的新的似然函数的最大值,与之前模型的似然函数的最大值进行比较。如果参数限制是正确的,那么加入这样一个参数应当不会造成似然函数最大值的大幅变动。一般使用两者的比例来进行比较,这个比值服从卡方分布。
1.2 R代码
这里用到了lmtest包中的lrtest()函数,使用R自带的数据集mtcars做演示
install.packages("lmtest")
library(lmtest)
mtcars
# mpg cyl disp hp drat wt qsec vs am gear carb
#Mazda RX4 21.0 6 160.0 110 3.90 2.620 16.46 0 1 4 4
#Mazda RX4 Wag 21.0 6 160.0 110 3.90 2.875 17.02 0 1 4 4
#Datsun 710 22.8 4 108.0 93 3.85 2.320 18.61 1 1 4 1
#Hornet 4 Drive 21.4 6 258.0 110 3.08 3.215 19.44 1 0 3 1
#Hornet Sportabout 18.7 8 360.0 175 3.15 3.440 17.02 0 0 3 2
#Valiant 18.1 6 225.0 105 2.76 3.460 20.22 1 0 3 1
#Duster 360 14.3 8 360.0 245 3.21 3.570 15.84 0 0 3 4
#Merc 240D 24.4 4 146.7 62 3.69 3.190 20.00 1 0 4 2
#Merc 230 22.8 4 140.8 95 3.92 3.150 22.90 1 0 4 2
#Merc 280 19.2 6 167.6 123 3.92 3.440 18.30 1 0 4 4
#Merc 280C 17.8 6 167.6 123 3.92 3.440 18.90 1 0 4 4
#Merc 450SE 16.4 8 275.8 180 3.07 4.070 17.40 0 0 3 3
#Merc 450SL 17.3 8 275.8 180 3.07 3.730 17.60 0 0 3 3
#Merc 450SLC 15.2 8 275.8 180 3.07 3.780 18.00 0 0 3 3
#Cadillac Fleetwood 10.4 8 472.0 205 2.93 5.250 17.98 0 0 3 4
#Lincoln Continental 10.4 8 460.0 215 3.00 5.424 17.82 0 0 3 4
#Chrysler Imperial 14.7 8 440.0 230 3.23 5.345 17.42 0 0 3 4
#Fiat 128 32.4 4 78.7 66 4.08 2.200 19.47 1 1 4 1
#Honda Civic 30.4 4 75.7 52 4.93 1.615 18.52 1 1 4 2
#Toyota Corolla 33.9 4 71.1 65 4.22 1.835 19.90 1 1 4 1
#Toyota Corona 21.5 4 120.1 97 3.70 2.465 20.01 1 0 3 1
#Dodge Challenger 15.5 8 318.0 150 2.76 3.520 16.87 0 0 3 2
#AMC Javelin 15.2 8 304.0 150 3.15 3.435 17.30 0 0 3 2
#Camaro Z28 13.3 8 350.0 245 3.73 3.840 15.41 0 0 3 4
#Pontiac Firebird 19.2 8 400.0 175 3.08 3.845 17.05 0 0 3 2
#Fiat X1-9 27.3 4 79.0 66 4.08 1.935 18.90 1 1 4 1
#Porsche 914-2 26.0 4 120.3 91 4.43 2.140 16.70 0 1 5 2
#Lotus Europa 30.4 4 95.1 113 3.77 1.513 16.90 1 1 5 2
#Ford Pantera L 15.8 8 351.0 264 4.22 3.170 14.50 0 1 5 4
#Ferrari Dino 19.7 6 145.0 175 3.62 2.770 15.50 0 1 5 6
#Maserati Bora 15.0 8 301.0 335 3.54 3.570 14.60 0 1 5 8
#Volvo 142E 21.4 4 121.0 109 4.11 2.780 18.60 1 1 4 2
model1 <- lm(mpg~cyl+disp+hp,data=mtcars)
#mpg=英里每加仑油,cyl=气缸数,disp=排量,hp=总马力,wt=车重
model2 <- lm(mpg~cyl+disp+hp+wt,data=mtcars)
lrtest(model1,model2)
结果:
Likelihood ratio test
Model 1: mpg ~ cyl + disp + hp
Model 2: mpg ~ cyl + disp + hp + wt
#Df LogLik Df Chisq Pr(>Chisq)
1 5 -79.009
2 6 -72.169 1 13.681 0.0002167 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1看到卡方检验的pvalue是显著的,认为model1和model2有差别,即加入的约束变量wt是一个在cyl,disp,hp作为协变量时对mpg有影响的变量。
2.eQTL分析
2.1 原理介绍
QTL(quantitative trait Loci),中文名是数量性状位点,是指生物体内和数量性状相关的遗传位点(大部分都是SNP,单核苷酸多态性)。而eQTL则是表达数量性状位点,是指基因表达量这个数量性状的相关遗传位点。
一般而言,eQTL主要分为两类:(1)顺式eQTL(cis-eQTL):它主要是指与所调控基因相距较近的eQTL,一般多位于所调控基因的上下游1Mb区域;(2)反式eQTL(trans-eQTL):与cis-eQTL恰恰相反,反式是指距离所调控基因位置比较远的eQTL,有时候距离甚至超过5Mb,甚至在不同染色体上。因此,对于eQTL分析而言,我们通常需要考虑两点,SNP和基因表达水平的关联度以及SNP与基因的距离。
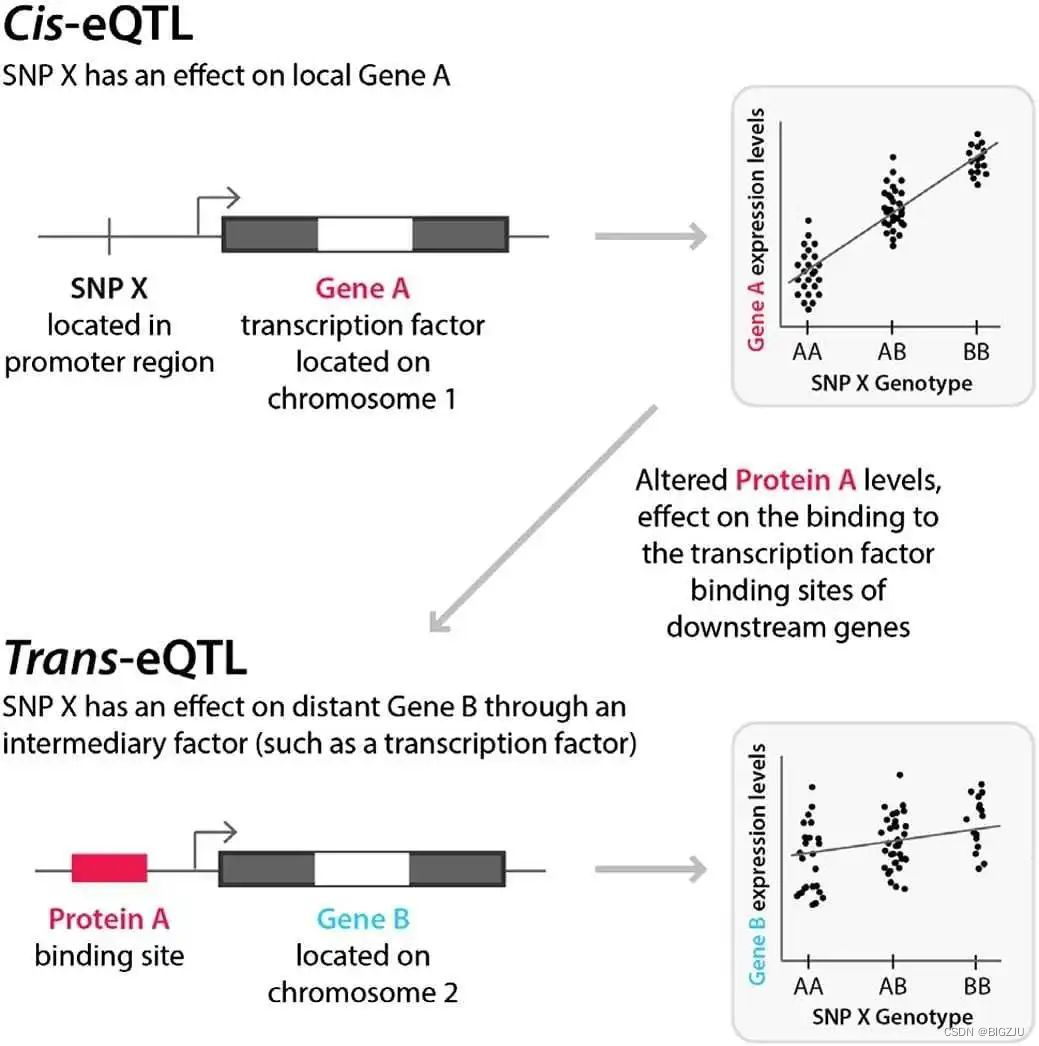
eQTL主要运用的是线性模型,
其中指的是基因的表达水平;
指的是SNP的基因类型,编码为0,1,2,分别表示主等位基因纯合AA、主次等位基因杂合AB、次等位基因纯和BB;
指的是AA基因型的表达水平;
指的是每一个等位基因B对Y的表达有多大的影响;
指的是随机误差。
以此来判断基因表达水平和SNPs之间的关系。
2.2 R代码
主要运用MatrixEQTL包的Matrix_eQTL_engine()函数进行eQTL分析,示范数据采用该包自带的数据
install.packages("MatrixEQTL") # 安装R包
library('MatrixEQTL')
##获取数据以及参数设置
base.dir = find.package("MatrixEQTL") # 获取该包的路径信息
useModel = modelLINEAR # 三种模型可选(modelANOVA or modelLINEAR or modelLINEAR_CROSS)
SNP_file_name = paste(base.dir, "/data/SNP.txt", sep="") # 获取SNP文件位置
SNP_file = data.table::fread(SNP_file_name, header=T) # 读取SNP文件,可以在R中查看,0纯合,1杂合,2次等位纯和
#SNP_file数据
# snpid Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10 Sam_11 Sam_12 Sam_13 Sam_14
# <char> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int>
# 1: Snp_01 2 0 2 0 2 1 2 1 1 1 2 2 1 2
# 2: Snp_02 0 1 1 2 2 1 0 0 0 1 1 1 1 0
# 3: Snp_03 1 0 1 0 1 1 1 1 0 1 1 0 1 1
# 4: Snp_04 0 1 2 2 2 1 1 0 0 0 1 2 1 1
# 5: Snp_05 1 1 2 1 1 2 1 1 0 1 1 2 0 1
# 6: Snp_06 2 2 2 1 1 0 1 0 2 1 1 1 2 0
# 7: Snp_07 1 1 2 2 0 1 1 1 1 0 2 2 0 1
# 8: Snp_08 1 0 1 0 1 0 0 1 1 1 0 2 0 1
# 9: Snp_09 2 1 2 2 0 1 1 0 2 1 1 0 1 1
# 10: Snp_10 1 1 0 0 0 2 2 1 1 2 1 1 1 1
# 11: Snp_11 2 2 2 0 2 1 1 2 1 2 0 1 0 1
# 12: Snp_12 1 1 2 2 2 1 1 1 1 0 2 0 1 1
# 13: Snp_13 0 1 1 1 1 1 2 1 2 2 0 0 0 1
# 14: Snp_14 0 0 1 0 1 2 2 2 1 1 1 0 1 0
# 15: Snp_15 1 2 1 2 2 1 2 1 1 2 1 1 1 2
# Sam_15 Sam_16
# <int> <int>
# 1: 2 1
# 2: 1 1
# 3: 1 2
# 4: 1 0
# 5: 2 1
# 6: 2 1
# 7: 1 1
# 8: 1 1
# 9: 0 0
# 10: 1 0
# 11: 1 2
# 12: 0 2
# 13: 1 1
# 14: 1 0
# 15: 0 2
expression_file_name = paste(base.dir, "/data/GE.txt", sep="") # 获取基因表达量文件位置
expression_file = data.table::fread(expression_file_name, header=T) # 读取基因表达量文件,可以在R中查看
#expression_file数据
# geneid Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10 Sam_11 Sam_12 Sam_13 Sam_14
# <char> <num> <num> <num> <num> <num> <num> <num> <num> <num> <num> <num> <num> <num> <num>
# 1: Gene_01 4.91 4.63 5.18 5.07 5.74 5.09 5.31 5.29 4.73 5.72 4.75 4.54 5.01 5.03
# 2: Gene_02 13.78 13.14 13.18 13.04 12.85 13.07 13.09 12.83 14.94 13.86 15.26 14.73 14.08 14.33
# 3: Gene_03 12.06 12.29 13.07 13.65 13.86 12.84 12.29 13.03 13.13 14.93 12.40 13.38 13.70 14.49
# 4: Gene_04 11.63 11.88 12.74 12.66 13.16 11.99 11.97 12.81 11.74 13.29 10.88 11.37 12.09 12.50
# 5: Gene_05 14.72 14.66 14.63 15.91 15.46 14.74 15.17 15.01 14.05 14.90 12.96 13.56 14.06 14.44
# 6: Gene_06 12.29 12.23 12.47 13.21 12.63 12.18 12.44 12.45 13.22 12.70 12.93 12.84 13.20 13.19
# 7: Gene_07 12.56 12.71 12.49 13.41 13.60 12.35 12.27 13.42 14.73 15.47 13.85 14.31 15.25 15.55
# 8: Gene_08 12.27 12.58 12.61 13.02 12.86 12.32 12.30 13.19 15.21 14.60 14.72 14.56 14.98 14.92
# 9: Gene_09 9.82 9.29 8.95 8.18 8.11 9.43 9.17 9.25 10.95 9.82 12.60 11.99 11.20 10.31
# 10: Gene_10 14.24 14.52 14.62 13.65 13.46 14.04 13.97 13.73 12.29 12.01 13.47 13.30 12.34 12.23
# Sam_15 Sam_16
# <num> <num>
# 1: 4.84 4.44
# 2: 14.72 14.97
# 3: 14.14 13.35
# 4: 11.40 11.45
# 5: 14.12 13.76
# 6: 12.64 12.80
# 7: 14.90 14.04
# 8: 14.53 14.72
# 9: 11.13 11.69
# 10: 12.80 12.47
covariates_file_name = paste(base.dir, "/data/Covariates.txt", sep="") # 读取协变量文件
covariates_file = data.table::fread(covariates_file_name, header=T) # 读取协变量文件,可在R中查看
#covariates数据
# id Sam_01 Sam_02 Sam_03 Sam_04 Sam_05 Sam_06 Sam_07 Sam_08 Sam_09 Sam_10 Sam_11 Sam_12 Sam_13 Sam_14
# <char> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int> <int>
# 1: gender 0 0 0 0 0 0 0 0 1 1 1 1 1 1
# 2: age 36 40 46 65 69 43 40 54 44 70 24 34 55 62
# Sam_15 Sam_16
# <int> <int>
# 1: 1 1
# 2: 48 40
output_file_name = tempfile() # 将输出文件设置为临时文件
pvOutputThreshold = 1e-2 # 定义gene-SNP associations的显著性P值0.01
errorCovariance = numeric() # 定义误差项的协方差矩阵 (用的很少)
snps = SlicedData$new() # 创建SNP文件为S4对象(S4对象是该包的默认输入方式)
snps$fileDelimiter = "\t" # 指定SNP文件的分隔符
snps$fileOmitCharacters = "NA" # 定义缺失值
snps$fileSkipRows = 1 # 跳过第一行(适用于第一行是列名的情况)
snps$fileSkipColumns = 1 # 跳过第一列(适用于第一列是SNP ID的情况
snps$fileSliceSize = 2000 # 每次读取2000条数据
snps$LoadFile( SNP_file_name ) # 载入SNP文件
# 下面的代码同snps的那部分一致
gene = SlicedData$new()
gene$fileDelimiter = "\t"
gene$fileOmitCharacters = "NA"
gene$fileSkipRows = 1
gene$fileSkipColumns = 1
gene$fileSliceSize = 2000
gene$LoadFile( expression_file_name )
cvrt = SlicedData$new()
cvrt$fileDelimiter = "\t"
cvrt$fileOmitCharacters = "NA"
cvrt$fileSkipRows = 1
cvrt$fileSkipColumns = 1
cvrt$fileSliceSize = 2000
cvrt$LoadFile( covariates_file_name )
# 文件的输入部分结束
me = Matrix_eQTL_engine( # 这是进行eQTL分析的主要函数
snps = snps, # 指定SNP 文件
gene = gene, # 指定基因表达量文件
cvrt = cvrt, # 指定协变量文件
output_file_name = output_file_name, # 指定输出文件
pvOutputThreshold = pvOutputThreshold, # 指定显著性P值
useModel = useModel, # 指定使用的计算模型
errorCovariance = errorCovariance, # 指定误差项的协方差矩阵
verbose = TRUE,#展示更多过程上的细节
pvalue.hist = TRUE,#记录p的分布
min.pv.by.genesnp = FALSE,#p值达不到阈值就不再记录。为False时运行的更快
noFDRsaveMemory = FALSE)#True时eqtl不记录在返回变量中,而直接在输出文件中。
res <- me$all$eqtls # 把eQTL的显著结果存储到变量res里
res # 查看结果
## snps gene statistic pvalue FDR beta
# 1 Snp_05 Gene_03 38.812160 5.515519e-14 8.273279e-12 0.4101317
# 2 Snp_13 Gene_09 -3.914403 2.055817e-03 1.541863e-01 -0.2978847
# 3 Snp_11 Gene_06 -3.221962 7.327756e-03 2.853368e-01 -0.2332470
# 4 Snp_04 Gene_10 3.201666 7.608981e-03 2.853368e-01 0.2321123
# 5 Snp_14 Gene_01 3.070005 9.716705e-03 2.915011e-01 0.2147077
#有5个和表达量相关的snps
#cis-eQTL和trans-eQTL
#相比普通的eQTL要额外读入snp和gene的位置文件
snps_location_file_name = paste(base.dir, "/data/snpsloc.txt", sep="")
gene_location_file_name = paste(base.dir, "/data/geneloc.txt", sep="")
snpspos = read.table(snps_location_file_name, header = TRUE, stringsAsFactors = FALSE)
#snpspos
# snpid chr pos
# 1 Snp_01 chr1 721289
# 2 Snp_02 chr1 752565
# 3 Snp_03 chr1 777121
# 4 Snp_04 chr1 785988
# 5 Snp_05 chr1 792479
# 6 Snp_06 chr1 798958
# 7 Snp_07 chr1 888658
# 8 Snp_08 chr1 918572
# 9 Snp_09 chr1 926430
# 10 Snp_10 chr1 947033
# 11 Snp_11 chr2 721289
# 12 Snp_12 chr2 752565
# 13 Snp_13 chr2 777121
# 14 Snp_14 chr2 785988
# 15 Snp_15 chr2 792479
genepos = read.table(gene_location_file_name, header = TRUE, stringsAsFactors = FALSE)
#genepos
# geneid chr left right
# 1 Gene_01 chr1 721289 731289
# 2 Gene_02 chr1 752565 762565
# 3 Gene_03 chr1 777121 787121
# 4 Gene_04 chr1 785988 795988
# 5 Gene_05 chr1 792479 802479
# 6 Gene_06 chr1 798958 808958
# 7 Gene_07 chr1 888658 898658
# 8 Gene_08 chr1 918572 928572
# 9 Gene_09 chr1 926430 936430
# 10 Gene_10 chr1 1000000 1010000
cisDist = 1e6#设置定义cis和trans的距离,这里是1Mb
output_file_name_cis = tempfile()
output_file_name_tra = tempfile()
pvOutputThreshold_cis = 1e-2
pvOutputThreshold_tra = 1e-2
me_ct = Matrix_eQTL_main(
snps = snps,
gene = gene,
cvrt = cvrt,
output_file_name = output_file_name_tra,
pvOutputThreshold = pvOutputThreshold_tra,
useModel = useModel,
errorCovariance = errorCovariance,
verbose = TRUE,
output_file_name.cis = output_file_name_cis,
pvOutputThreshold.cis = pvOutputThreshold_cis,
snpspos = snpspos,
genepos = genepos,
cisDist = cisDist,
pvalue.hist = TRUE,
min.pv.by.genesnp = FALSE,
noFDRsaveMemory = FALSE)
show(me_ct$cis$eqtls)#展示cis-eQTL
# snps gene statistic pvalue FDR beta
# 1 Snp_05 Gene_03 38.812160 5.515519e-14 5.515519e-12 0.4101317
# 2 Snp_04 Gene_10 3.201666 7.608981e-03 3.804491e-01 0.2321123
#2个snp是cis-eQTL
show(me_ct$trans$eqtls)#展示trans-eQTL
# snps gene statistic pvalue FDR beta
# 1 Snp_13 Gene_09 -3.914403 0.002055817 0.1027908 -0.2978847
# 2 Snp_11 Gene_06 -3.221962 0.007327756 0.1619451 -0.2332470
# 3 Snp_14 Gene_01 3.070005 0.009716705 0.1619451 0.2147077
#3个是trans-eQTL

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









