






背景知识
Illumina测序技术,即边合成边测序(SBS)技术,是全球广泛采用的新一代测序(NGS)技术,负责生成全球90%以上的测序数据。随着每个 dNTPs 的加入,荧光标记的可逆终止子被成像,然后切割以允许下一个碱基的掺入。由于所有 4 个可逆终止子结合的 dNTPs 在每个测序循环中都存在,将偏差降至最低。该方法实际上消除了与重复核苷酸(均聚物)串相关的错误和漏检。Illumina 技术支持单端和双端文库(即单端测序or双端测序)。SBS技术提供了用于高分辨率基因组测序的短插入双端功能,以及用于高效序列组装、从头测序等的长插入双端读取。短插入片段和长读段的结合提高了对任何基因组进行全面表征的能力。
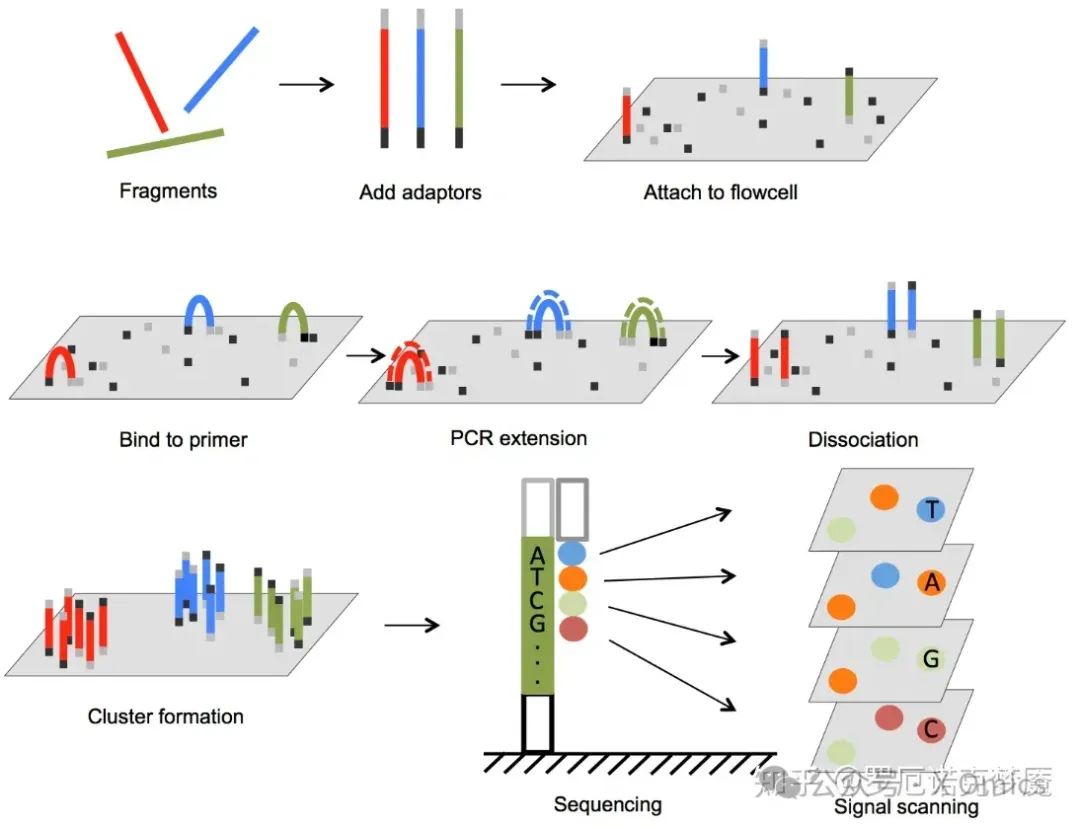
被称为下一代测序 (NGS) 的大规模并行测序技术已经彻底改变了生物科学。凭借其超高的吞吐量、可扩展性和速度,NGS 使研究人员能够以前所未有的水平执行各种应用和研究生物系统。例如,NGS 允许研究人员:
-
- 快速测序全基因组
-
- 放大到深度序列目标区域
-
- 利用 RNA-Seq 发现新的 RNA 变体和剪接位点,或量化 mRNA 以进行基因表达分析
-
- 分析表观遗传因素,例如全基因组 DNA 甲基化和 DNA-蛋白质相互作用
-
- 对癌症样本进行测序以研究罕见的体细胞变异、肿瘤亚克隆等研究人类或环境中的微生物多样性

RNA-seq工作流程主要分为以下三步:

-
1. 文库制备 。
-
2. 测序。
-
3. 数据分析。
分析流程(Analysis Pipeline)
上游分析的过程需要在Linux系统中完成。由上述测序技术所得到的原始测序文件为.fastq格式文件,其主要格式为:
@A00184:675:HKHGGDSXY:2:1101:1181:1000 1:N:0:AGTGGCTA+CCAAGGATCCTCCATCAGGTATTGCTCCAGGGACACTGGGTGCTTGATGTAGACATTGGTCTGTATGTCCTTGGCAGGCAGCCGCTCCAACTCCGTGTGGAACTCAGCCACCCGGTTCTGGGACAGCAGGAAGAGGAGGTTGAGGCCCAAGAGCTGGT+,::FFF:::FFFFFFF:F:F:FFFF:F:FFFFFF:FFFFFFFFFFFFFFFFF:FFFFFFFF,FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,F:FFFFF::FFFFF:FFFFFFFFFFFF,,FFFFFFFFFFFFFF:FFFFFFFFFFF@A00184:675:HKHGGDSXY:2:1101:1615:1000 1:N:0:AGTGGCTA+CCAAGGATAGGAGGACGACGGACGGACGGACGGACGGGCCGCGGACGGGCGGACGGGAGGGAGCGAGCGGGCGCGGGGGCGGCGGCCGGGACCGGTGGGGCCGGGGCGGGGCGCGGCGAACCGGACGCCCCAACCACCCGCCCCCCCCCCGCCACCAC+:FFFFF:FFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFFF,F:FF,FF,FFFFFF,:,F,,F::,FF
第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份.fastq文件中不会重复出现,甚至不同的.fastq文件里也不会有重复;
第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基(illumina测序的接头);
第三行:以‘+’开头,在旧版的.fastq文件中会直接重复第一行的信息,但现在一般什么也不加;
第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。https://zhuanlan.zhihu.com/p/382389814
1.准备工作(参考基因组和基因组注释文件)常用UCSC或Ensembl数据库
1)参考基因组(.fa.gz)的下载。实验室常用用物种基因组:
-
Human genome(GRCh38/hg38):Index of /goldenPath/hg38/chromosomes (ucsc.edu)
-
Mouse genome (GRCm39/mm39):Index of /goldenPath/mm39/bigZips (ucsc.edu)
-
D. melanogaster genome(BDGP Release 6 + ISO1 MT/dm6):Index of /goldenPath/dm6/bigZips (ucsc.edu)
2)注释文件(.gtf/gff)下载
-
Human genome Annotation: Index of /pub/release-104/gtf/homo_sapiens/ (ensembl.org)
-
Mouse genome:Index of /pub/release-104/gtf/mus_musculus/ (ensembl.org)
-
D. melanogaster genome:Index of /pub/release-104/gtf/drosophila_melanogaster/ (ensembl.org)
## 下载命令(基于linux)axel <下载连接> #orwget -c <下载连接>## 解压命令(基于linux)gunzip <.gtf.gz文件>#.gtf文件使用前需要解压缩,参考基因组(.fa.gz文件)不用
2.质量控制
Trim Galore是对FastQC和Cutadapt的包装。适用于所有高通量测序,包括RRBS(Reduced Representation Bisulfite-Seq),Illumina、Nextera 和smallRNA测序平台的双端和单端数据。该软件会对数据进行以下4步处理:
①去除reads 3’端的低质量碱基:illumina平台的测序数据,通常3’端质量较差。trim_galore首先会过滤掉3’端的低质量碱基,本质上是调用了cutadapt的质量过滤算法。下图是过滤前后碱基质量的分布图,可以看到,过滤掉低质量碱基后,序列的整体质量显著提高。
②去除adapter序列:过滤掉低质量的碱基之后,trim_galore会调用cutadapt在reads的3’端查找adapter 序列并去除。通常情况下,我们需要指定对应的adapter序列,如果没有指定的化,trim_galore会自动查找以下3种类型的adapter:
Illumina: AGATCGGAAGAGC
Small RNA: TGGAATTCTCGG
Nextera: CTGTCTCTTATA默认读取前一百万条序列,通过这一百万条序列判断adapter属于上述三种的哪一种,然后进行去除。如果你不希望软件自动判断,也可以通过--illumina, --nextera, --small_rna参数指定对应的adapter类型。
③去除长度太短的序列:经过上述两步处理之后,有可能剩余的序列长度很短,这部分短序列也会被去除。默认情况下,如果序列长度少于20bp, 这条序列会被丢掉。
④其它过滤:对于所有的输入序列,以上3个步骤是肯定会执行的。除此之,trim_galore还支持一些其他的过滤措施,以满足个性化的需求。
hardtrim5参数用于从序列的3’端切除碱基,示意如下
before: CCTAAGGAAACAAGTACACTCCACACATGCATA
--hardtrim5 20: CCTAAGGAAACAAGTACACT通过hardtrim5参数可以将序列截取成固定长度。与之对应的,还有一个hardtrim3参数,从序列的5’端切除碱基,示意如下
before: CAAATGTTATTTTTAAGAAAATGGAAAAT
--hardtrim3 20: TTTTTAAGAAAATGGAAAAT软件使用:
trim_galore -q 20 --phred33 --stringency 3 --length 20 -e 0.1 \--paired $dir/cmp/01raw_data/$fq1 $dir/cmp/01raw_data/$fq2 \--gzip -o $input_data
--quality:设定Phred quality score阈值,默认为20。
--phred33:选择-phred33或者-phred64,表示测序平台使用的Phred quality score。
--adapter:输入adapter序列。也可以不输入,Trim Galore!会自动寻找可能性最高的平台对应的adapter。自动搜选的平台三个,也直接显式输入这三种平台,即--illumina、--nextera和--small_rna。
--stringency:设定可以忍受的前后adapter重叠的碱基数,默认为1。可以适度放宽,因为后一个adapter几乎不可能被测序仪读到。
--length:设定输出reads长度阈值,小于设定值会被抛弃。
--paired:对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准。
--retain_unpaired:对于双端测序结果,一对reads中,如果一个read达到标准,但是对应的另一个要被抛弃,达到标准的read会被单独保存为一个文件。
--gzip和--dont_gzip:清洗后的数据zip打包或者不打包。
--output_dir:输入目录。需要提前建立目录,否则运行会报错。
-- trim-n : 移除read一端的reads。
3.序列比对
1)在进行reads比对前,我们需要先构建基因组索引。
## 构建基因组索引STAR --runThreadN 6 --runMode genomeGenerate \--genomeDir index_dir \--genomeFastaFiles genome.fasta \--sjdbGTFfile genome.gtf --sjdbOverhang 149
--runThreadN:线程数。--runMode genomeGenerate:构建基因组索引。--genomeDir:索引目录。(index_dir一定要是存在的文件夹,需提前建好)--genomeFastaFiles:基因组文件。--sjdbGTFfile:基因组注释文件。--sjdbOverhang:reads长度减1。
2)有了索引后,我们就可以进行reads比对了。
## 进行 reads 比对STAR --twopassMode Basic \--quantMode TranscriptomeSAM GeneCounts \--runThreadN 6 --genomeDir index_dir \--alignIntronMin 20 --alignIntronMax 50000 \--outSAMtype BAM SortedByCoordinate \--sjdbOverhang 149 \--outSAMattrRGline ID:sample SM:sample PL:ILLUMINA \--outFilterMismatchNmax 2 --outSJfilterReads Unique \--outSAMmultNmax 1 --outFileNamePrefix out_prefix \--outSAMmapqUnique 60 --readFilesCommand gunzip -c \--readFilesIn seq1.fq.gz seq2.fq.gz
--twopassMode Basic:使用two-pass模式进行reads比对。简单来说就是先按索引进行第一次比对,而后把第一次比对发现的新剪切位点信息加入到索引中进行第二次比对。
--quantMode TranscriptomeSAM GeneCounts:将reads比对至转录本序列。
--runThreadN:线程数。--genomeDir:索引目录。
--alignIntronMin:最短的内含子长度。(根据GTF文件计算)
--alignIntronMax:最长的内含子长度。(根据GTF文件计算)
--outSAMtype BAM SortedByCoordinate:输出BAM文件并进行排序。
--sjdbOverhang:reads长度减1。
--outSAMattrRGline:ID代表样本ID,SM代表样本名称,PL为测序平台。在使用GATK进行SNP Calling时同一SM的样本可以合并在一起。
--outFilterMismatchNmax:比对时允许的最大错配数。
--outSJfilterReads Unique:对于跨越剪切位点的reads(junction reads),只考虑跨越唯一剪切位点的reads。
--outSAMmultNmax:每条reads输出比对结果的数量。
--outFileNamePrefix:输出文件前缀。
--outSAMmapqUnique 60:将uniquely mapping reads的MAPQ值调整为60,满足下游使用GATK进行分析的需要。
--readFilesCommand:对FASTQ文件进行操作。
--readFilesIn:输入FASTQ文件的路径。
4.表达定量
featureCounts 需要两个输入文件:比对产生的BAM/ SAM文件、区间注释文件。对于区间文件而言,支持以下两种格式:GTF 格式、SAF 格式。
features 支持对单个样本定量,还支持对多个样本进行归一化。单个样本定量的用法如下:
featureCounts -T 5 -t exon -g gene_id \-a annotation.gtf \-o counts.txt mapping.sam
多个样本归一化的用法如下:
featureCounts -t exon -g gene_id -a annotation.gtf \-o counts.txt library1.bam \library2.bam library3.bam
-a参数指定的区间注释文件,默认是gtf格式。
-T参数指定线程数,默认是1。-t参数指定想要统计的feature的名称,取值范围是gtf 文件中的第3列的值,默认是exon。
-g参数指定想要统计的meta-feature的名称,取值范围参考gtf第9列注释信息,gtf的第9列为key=value的格式,-g参数可能的取值就是所有的key, 默认值是gene_id。
最后将得到的定量结果文件提取处理,获得最后的定量结果文件:
awk 'BEGIN{FS=OFS="\t"}!/#/{print $1,$6,$7}' repeats.count/${sample}.repeats.count >repeats.count/${sample}.repeats.count.simple结果如下(三列为:基因名称,长度及定量结果):
Geneid Length starOut/sample.coding_genes.Aligned.sortedByCoord.out.bamC1orf141 2533 0PKP1 5447 0RERE 8321 712CSMD2 13920 3HIVEP3 14115 15SLC44A5 4799 271TTLL7 8052 803MAN1A2 8590 2619GLMN 2032 960
根据表达定量的结果就可以进行下游分析,找到不同样本之间基因表达量的差异或作用关系,为下一步的研究指明方向。
5.数据标准化
下游分析需要在R中完成。需要安装R包:DESeq2、ggplot2。
定量结果文件读入:
## R包的下载及调用if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")BiocManager::install("DESeq2")BiocManager::install("ggplot2")suppressMessages(library(DESeq2))suppressMessages(library(ggplot2))## 输入定量结果文件filelist<-dir("<定量结果文件夹/>",pattern = "<定量结果文件后缀—eg. *simple>")filelist <- paste("<定量结果文件夹/>",filelist,sep="")file1.count<-read.table(filelist[1],header = T,sep = "\t")count<-matrix(ncol = length(filelist)+1,nrow = nrow(file1.count))count<-as.data.frame(count)count[,1:2]<-file1.count[,c(1,3)]for(i in 2:length(filelist)){file.count<-read.table(filelist[i],header = T,sep = "\t")print(filelist[i])count[,i+1]<-file.count[,3]}head(count)colnames(count) <- c("Geneid",filelist)rownames(count) <- count$Geneidcount <- count[,-1]
6.主成分分析及差异分析
colData <- data.frame(row.names =colnames(count),ALT.group=pheno$ALT.group)dds <- DESeqDataSetFromMatrix(countData = count, colData = colData, design = ~ ALT.group)cpm <- fpm(dds,robust = FALSE)cpm <- as.data.frame(cpm)vst <- vst(dds)plotPCA(vst,intgroup="group_name")plotPCA(vst,intgroup="group_name ")data <- plotPCA(vst,intgroup="ALT.group",returnData = TRUE)data$cell.name <- rownames(data)data.pheno <- merge(data,pheno,by="cell.name")data.phenoggplot(data.pheno, aes(x = PC1, y=PC2)) +geom_point(size=4.5,stroke = 0.8,aes(shape=origin,color=group )) +labs(title=" ") +theme(plot.title=element_text(hjust=0.5),title =element_text(size=12) ) +xlab(paste0( " PC1: " ," * % variance " ))+ylab(paste0( " PC2: " , " * % variance " ))dds <- DESeq(dds)res <- results(dds,contrast = c("样本类型","type A","type B"))res <- as.data.frame(res)cpm.res <- merge(cpm,res,by="repeatMasker.repName")cpm.res.anno <- merge(anno,cpm.res,by="repeatMasker.repName")write.csv(cpm.res.anno,file="<输出文件名>")
7.结果可视化
1)火山图
suppressMessages(library(ggplot2))suppressMessages(library(ggrepel))rm(list=ls())dataset <- read.csv("sample.csv",header = T)# 设置pvalue和logFC的阈值cut_off_pvalue = 0.05cut_off_logFC = 1# 根据阈值分别为上调基因设置‘up’,下调基因设置‘Down’,无差异设置‘Stable’,保存到change列# 这里的change列用来设置火山图点的颜色dataset$change = ifelse(dataset$pvalue < cut_off_pvalue & abs(dataset$log2FoldChange) >= cut_off_logFC,ifelse(dataset$log2FoldChange> cut_off_logFC ,'Up','Down'), 'Stable')# 绘制火山图====================================p <- ggplot(#设置数据dataset,aes(x = log2FoldChange, y = -log10(pvalue), colour=change)) +geom_point(alpha=0.4, size=3.5) +scale_color_manual(values=c("#546de5", "#d2dae2","#ff4757"))+# 辅助线geom_vline(xintercept=c(-1,1),lty=4,col="black",lwd=0.8) +geom_hline(yintercept = -log10(cut_off_pvalue),lty=4,col="black",lwd=0.8) +# 坐标轴labs(x="log2(fold change)",y="-log10 (p-value)")+theme_bw()+# 图例theme(plot.title = element_text(hjust = 0.5), legend.position="right", legend.title = element_blank())# 这里设置logFC值大于5的差异基因来标记# !!!需要注意的是标记的基因不能太多,Rstudio容易卡死dataset$label = ifelse(dataset$pvalue < cut_off_pvalue & abs(dataset$log2FoldChange) >= 2, as.character(dataset$gene),"")p + geom_text_repel(data = dataset, aes(x = dataset$log2FoldChange, y = -log10(dataset$pvalue), label = label),size = 3,box.padding = unit(0.5, "lines"),point.padding = unit(0.8, "lines"), segment.color = "black", show.legend = FALSE)
2)热图
pheatmap::pheatmap(data,cluster_rows = F,border_color = "grey",angle_col = 45,color = colorRampPalette(c("skyblue","white","pink"))(100))
3)KEGG分析和GO分析
得到差异分析结果后,筛选想要分析的gene,获得gene list并复制到输入框中进行分析。KEGG和GO分析都可以用DAVID在线工具完成。
library(ggplot2)library(dplyr)library(stringi)#BiocManager::install("AnnotationHub")rm(list = ls())options(stringsAsFactors = F)##DAVID富集结果可视化downgokegg=read.table("944_go.txt",sep = "\t",header = T)##数据输入及预处理enrich=downgokegg##只要数据格式是DAVID富集结果的格式,下面的代码可以全选→run,一键出图enrich_signif=enrich[which(enrich$PValue<0.001),]enrich_signif=enrich_signif[,c(1:3,5)]enrich_signif=data.frame(enrich_signif)GO=enrich_signif[which(enrich_signif$Category!="KEGG_PATHWAY"),]#KEGG=enrich_signif[which(enrich_signif$Category=="KEGG_PATHWAY"),]##处理TermGO$Term<-stri_sub(GO$Term,12,100) ##去掉编号#KEGG$Term<-stri_sub(KEGG$Term,10,100)##合并数据##排序,并取top5GO_BP=GO[which(GO$Category=="GOTERM_BP_DIRECT"),]GO_BP=arrange(GO_BP,GO_BP$PValue)[1:10,]GO_CC=GO[which(GO$Category=="GOTERM_CC_DIRECT"),]GO_CC=arrange(GO_CC,GO_CC$PValue)[1:10,]GO_MF=GO[which(GO$Category=="GOTERM_MF_DIRECT"),]GO_MF=arrange(GO_MF,GO_MF$PValue)[1:10,]#KEGG=arrange(KEGG,KEGG$PValue)[1:10,]##合并数据enrich_signif=rbind(GO_BP,rbind(GO_CC,rbind(GO_MF,KEGG)))######go=enrich_signifgo=arrange(go,go$Category,go$PValue)##图例名称设置m=go$Categorym=gsub("TERM","",m)m=gsub("_DIRECT","",m)go$Category=m##GO_term_order=factor(as.integer(rownames(go)),labels = go$Term)COLS<-c("#66C3A5","#8DA1CB","#FD8D62","red")##ggplot(data=go,aes(x=GO_term_order,y=Count,fill=Category))+geom_bar(stat = "identity",width = 0.8)+scale_fill_manual(values = COLS)+theme_bw()+xlab("Terms")+ylab("Gene_counts")+labs()+theme(axis.text.x = element_text(face = "bold",color = "gray50",angle = 45,vjust = 1,hjust = 1))
免责声明:本号对所有原创、转载文章陈述与观点均保持中立,内容仅供读者学习和交流。文章、图片等版权归原作者享有,如有侵权,请留言联系更正或删除。















 1465
1465

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









