






在第五次因为偷懒想使用本地RStudio跑scRNA-seq数据结果电脑死机蓝屏只能强制重启后——
终于开始配置linux系统服务器下的R了。
本文参考大佬教程:
https://blog.csdn.net/liangjinghui123/article/details/131494540
https://blog.csdn.net/luofunong/article/details/132870880
前情提要:服务器无sudo权限,无法修改默认目录以及各种Path
##linux配置R环境
##查看已有环境
conda info --envs
##新建conda环境
conda create -n R_temporary中途选择默认yes

##激活环境
source activate R_temporary
##添加channels
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
conda config --show
##查看R版本信息
conda search r-base
##选择合适的版本安装
conda install r-base=4.3.2全都选择y
安装成功后(done)进入R:
##进入R
R
##退出R
#q()先设置镜像源:
##查看镜像
options()$repos
# CRAN
#"@CRAN@"
##打开文件
file.edit(file.path("~",".Rprofile"))
##“a”进入编辑状态,写入代码,Esc结束编辑,“:wq”退出
#options("repos"= c(CRAN="https://mirrors.pku.edu.cn/CRAN/","http://mirrors.aliyun.com/CRAN"))
options("repos"=c(CRAN="https://mirrors.pku.edu.cn/CRAN/","https://mirrors.tuna.tsinghua.edu.cn/CRAN/","http://mirrors.aliyun.com/CRAN","https://mirrors.ustc.edu.cn/CRAN/"))
print("已设置北大阿里云镜像")
##退出再重新进入R,查看镜像是否设置好
q()
##进入R
R
##查看镜像
options()$repos
#保证镜像源设置好,装包才能顺利然后就开始在R中安装BiocManager:
##安装BiocManager:https://www.bioconductor.org/install/
#The current release of Bioconductor is version 3.18; it works with R version 4.3.0.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.18")在安装BiocManager时也容易遇到千奇百怪的报错:
URL 'https://bioconductor.org/config.yaml': status was 'Problem with the SSL CA cert (path? access rights?)'
还有:
Bioconductor version cannot be validated; no internet connection? See
#troubleshooting section in vignette
Warning messages:
1: In file(con, "r") :
URL 'https://bioconductor.org/config.yaml': status was 'Couldn't resolve host name'
2: In file(con, "r") :
URL 'http://bioconductor.org/config.yaml': status was 'Couldn't resolve host name'
Error: Bioconductor version cannot be validated; no internet connection? See
#troubleshooting section in vignette
一直报错,无法解决——
试试这样:https://cloud.tencent.com/developer/article/2228246
##让Bioconductor通过联网去验证版本
options(BIOCONDUCTOR_ONLINE_VERSION_DIAGNOSIS=TRUE)
install.packages("BiocManager")
library(BiocManager)
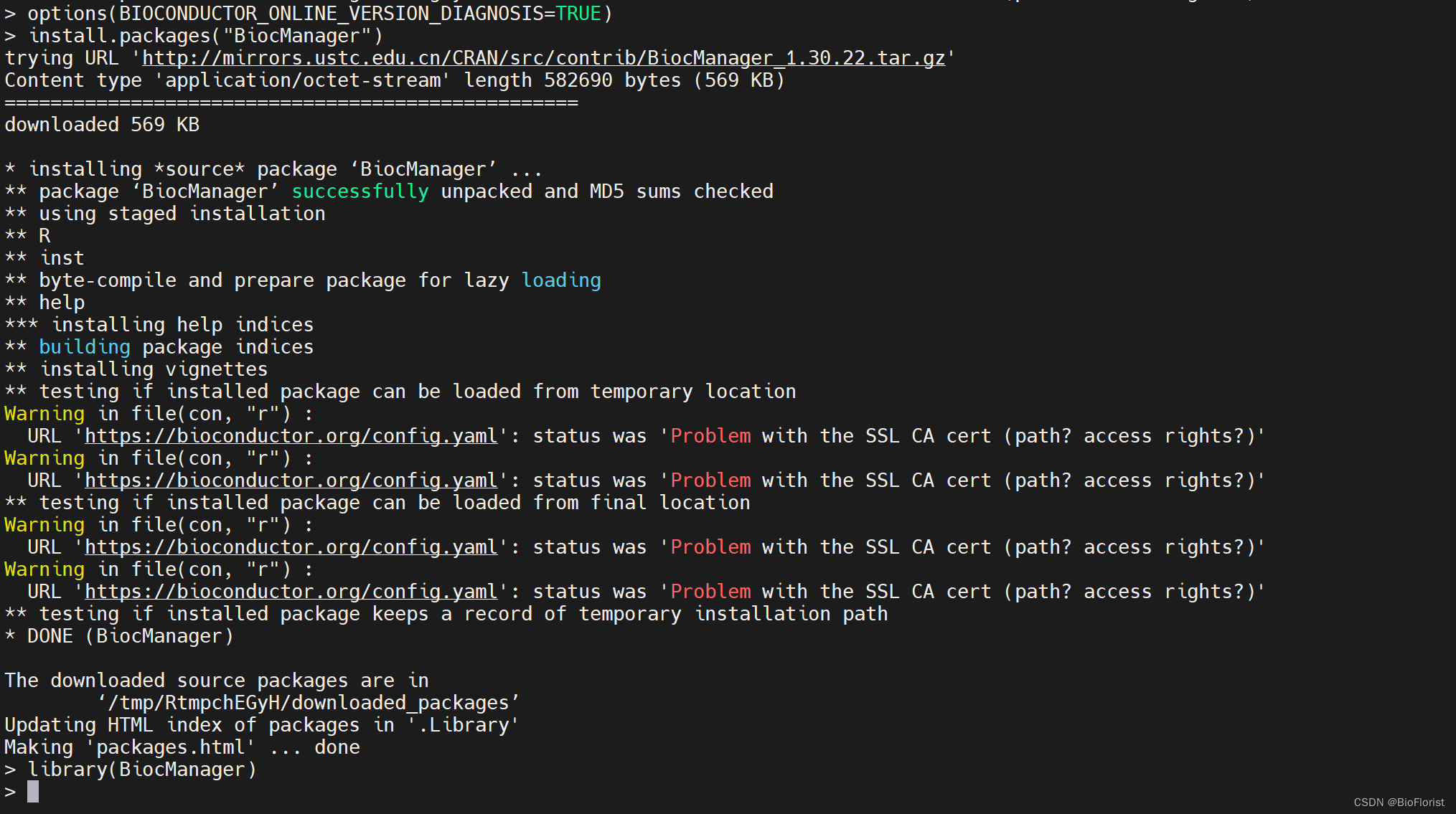
(其实完全没有装好,但是我装不下去了==)
继续——
正常安装R包:
install.packages("Seurat")#没装上
#version = 5.0.1
library(Seurat)
install.packages("tidyverse")#没装上
library(tidyverse)
install.packages("patchwork")
library(patchwork)
install.packages("ggplot2")
library(ggplot2)
install.packages("clustree")#没装上
library(clustree)
install.packages("cowplot")
library(cowplot)
install.packages("dplyr")
library(dplyr)
install.packages("harmony")
library(harmony)
BiocManager::install("monocle")#没装上
library(monocle)可以看到,我一共想安装9个R包,最终安装成功并能顺利library调用的只有5个!!!
经过我千奇百怪的搜索和安装尝试后,报错也千奇百怪:
make: *** [/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/etc/Makeconf:193: vendor/cigraph/src/community/optimal_modularity.o] Error 1
ERROR: compilation failed for package ‘igraph’
* removing ‘/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/library/igraph’
Warning message:
In install.packages("igraph") :
installation of package ‘igraph’ had non-zero exit status
Warning: unable to access index for repository https://mirrors.pku.edu.cn/CRAN/src/contrib:
cannot open URL 'https://mirrors.pku.edu.cn/CRAN/src/contrib/PACKAGES'
Warning: unable to access index for repository https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/contrib:
cannot open URL 'https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/contrib/PACKAGES'
Warning: dependency ‘graph’ is not available
甚至还有缺少编译文件:
In file included from vendor/cigraph/src/community/optimal_modularity.c:32:
vendor/cigraph/src/internal/glpk_support.h:39:10: fatal error: glpk.h: No such file or directory
39 | #include <glpk.h>
| ^~~~~~~~
compilation terminated.
make: *** [/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/etc/Makeconf:193: vendor/cigraph/src/community/optimal_modularity.o] Error 1
或者又是缺少依赖库:
Configuration failed to find the harfbuzz freetype2 fribidi library. Try installing:
* deb: libharfbuzz-dev libfribidi-dev (Debian, Ubuntu, etc)
* rpm: harfbuzz-devel fribidi-devel (Fedora, EPEL)
* csw: libharfbuzz_dev libfribidi_dev (Solaris)
* brew: harfbuzz fribidi (OSX)
If harfbuzz freetype2 fribidi is already installed, check that 'pkg-config' is in your
PATH and PKG_CONFIG_PATH contains a harfbuzz freetype2 fribidi.pc file. If pkg-config
is unavailable you can set INCLUDE_DIR and LIB_DIR manually via:
R CMD INSTALL --configure-vars='INCLUDE_DIR=... LIB_DIR=...'
-------------------------- [ERROR MESSAGE] ---------------------------
<stdin>:1:10: fatal error: hb-ft.h: No such file or directory
compilation terminated.
--------------------------------------------------------------------
ERROR: configuration failed for package ‘textshaping’
/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/bin/../lib/gcc/x86_64-conda-linux-gnu/13.2.0/../../../../x86_64-conda-linux-gnu/bin/ld: cannot find -lxml2: No such file or directory
collect2: error: ld returned 1 exit status
make: *** [/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/share/make/shlib.mk:10: xml2.so] Error 1
ERROR: compilation failed for package ‘xml2’
* removing
但没有sudo权限,根本无法补装依赖库……
在焦虑的驱使下,找到了这篇文章:
https://zhuanlan.zhihu.com/p/585291878
“退出R,使用conda安装”
本小白第一次知道R包还可以用conda安装……
直接开装最难搞也是最有用的Seurat包:
只需静静等待它成功:
进R试试:
注意了,Seurat包首字母可要大写啊!
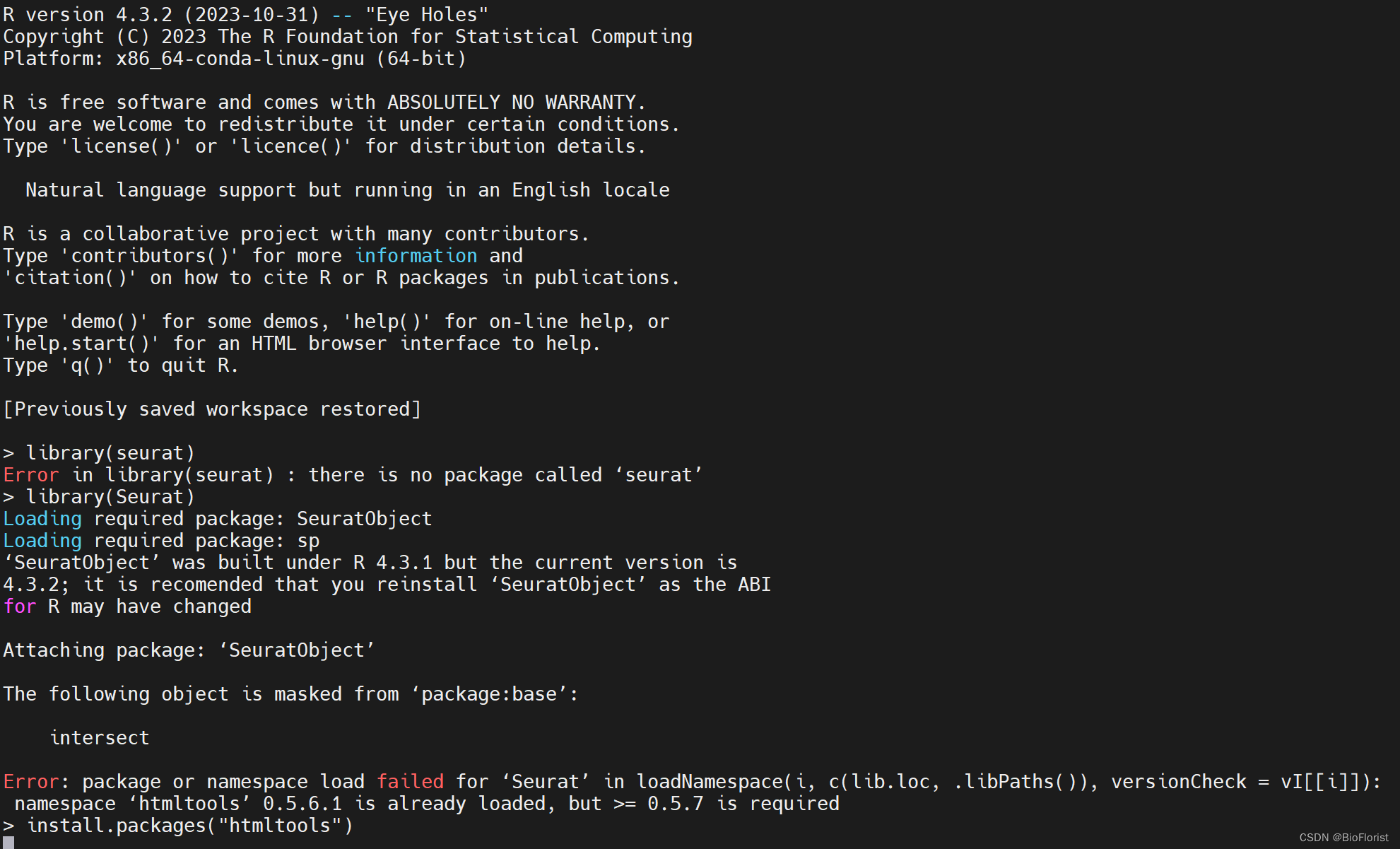
好好好,又报错:
Error: package or namespace load failed for ‘Seurat’ in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
namespace ‘htmltools’ 0.5.6.1 is already loaded, but >= 0.5.7 is required
install一下试试看呢?
install.packages("htmltools")Error: package or namespace load failed for ‘Seurat’ in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
namespace ‘htmltools’ 0.5.6.1 is already loaded, but >= 0.5.7 is required
仍然报版本错误……那把R包安装路径里的文件夹删掉再重新装呢?
#查看R包安装路径
.libPaths()
#去路径里把htmltools包文件夹删掉
#再次安装
install.packages("htmltools")继续报错:
ERROR: failed to lock directory ‘/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/library’ for modifying
Try removing ‘/hpc/data/home/xxx/software/miniconda3/envs/R_temporary/lib/R/library/00LOCK-htmltools’
再去路径里把00LOCK-htmltools文件夹删掉!
(如果删不掉就重新进入R再删)
删掉后再次安装:
install.packages("htmltools")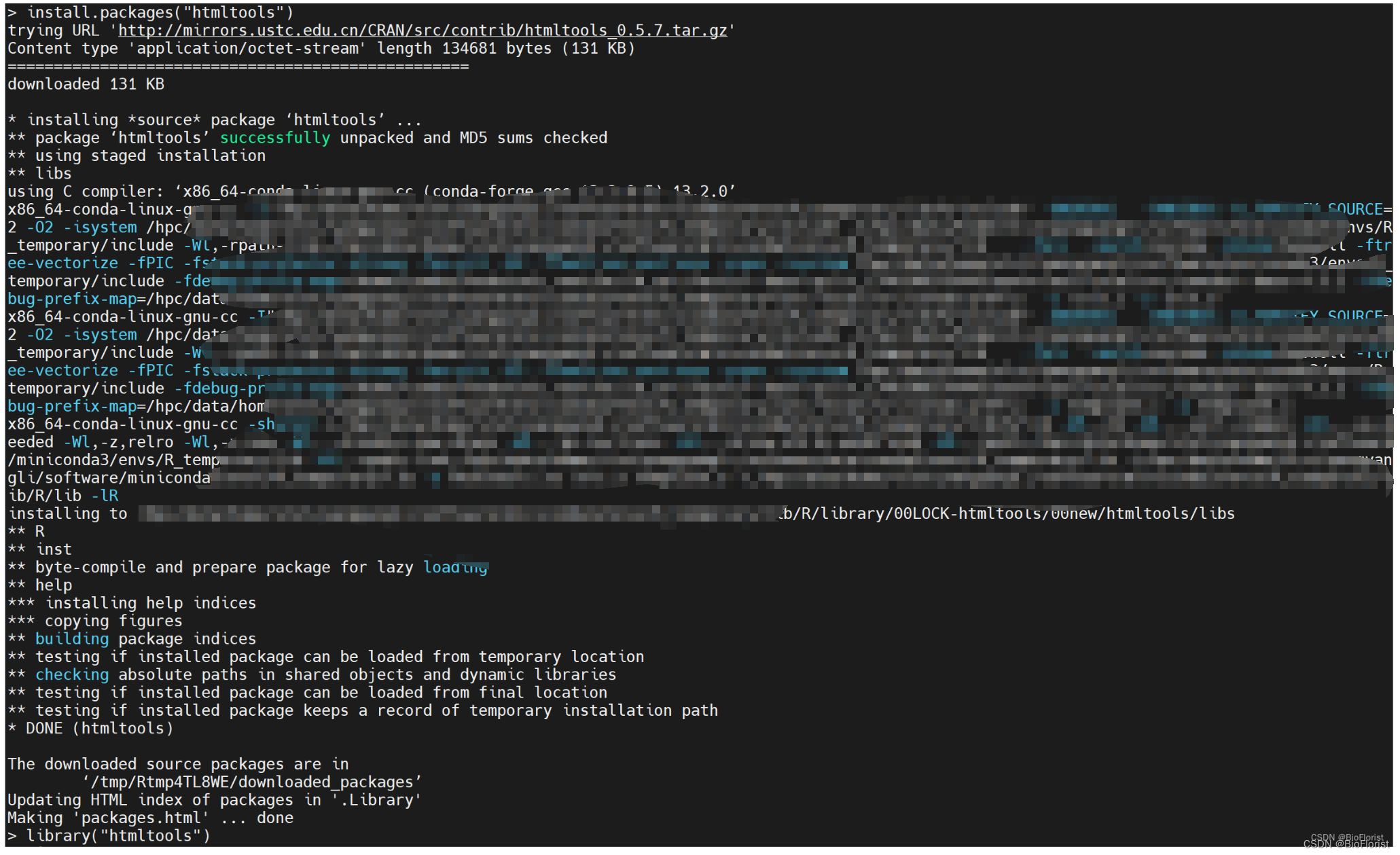
然后就很顺利地安装好啦~
再次调用Seurat:(== 你还记得安装htmltools是为了Seurat吗((
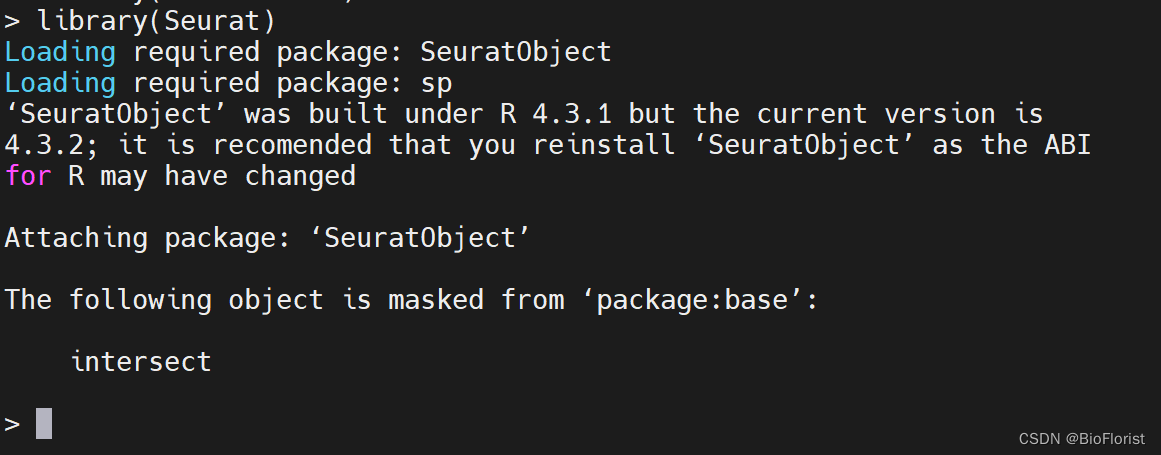
成功~
同理,tidyverse和clustree都可以成功安装~
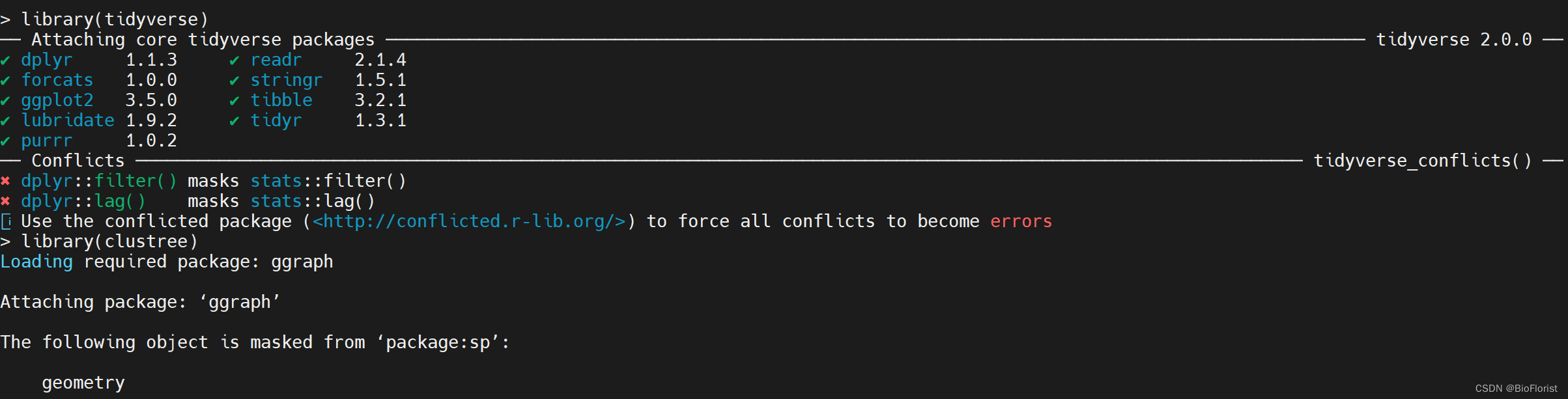
最后看看需要BiocManager安装的monocle:
也找到了大佬的trouble shooting:https://blog.csdn.net/wangyiqi806643897/article/details/125077367
运用一些普适性命令(别搞特殊(
在Bioconda上找到要安装的包的对应名字:
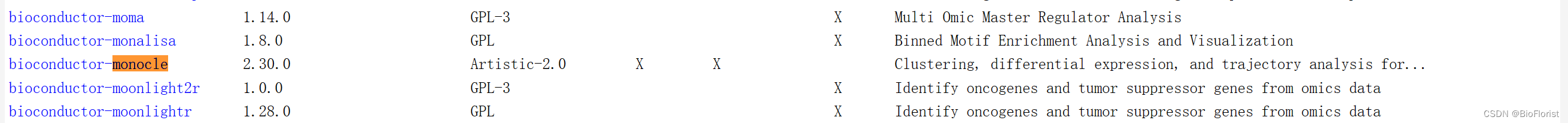
conda install -c bioconda bioconductor-monocle好好好,又报错了:
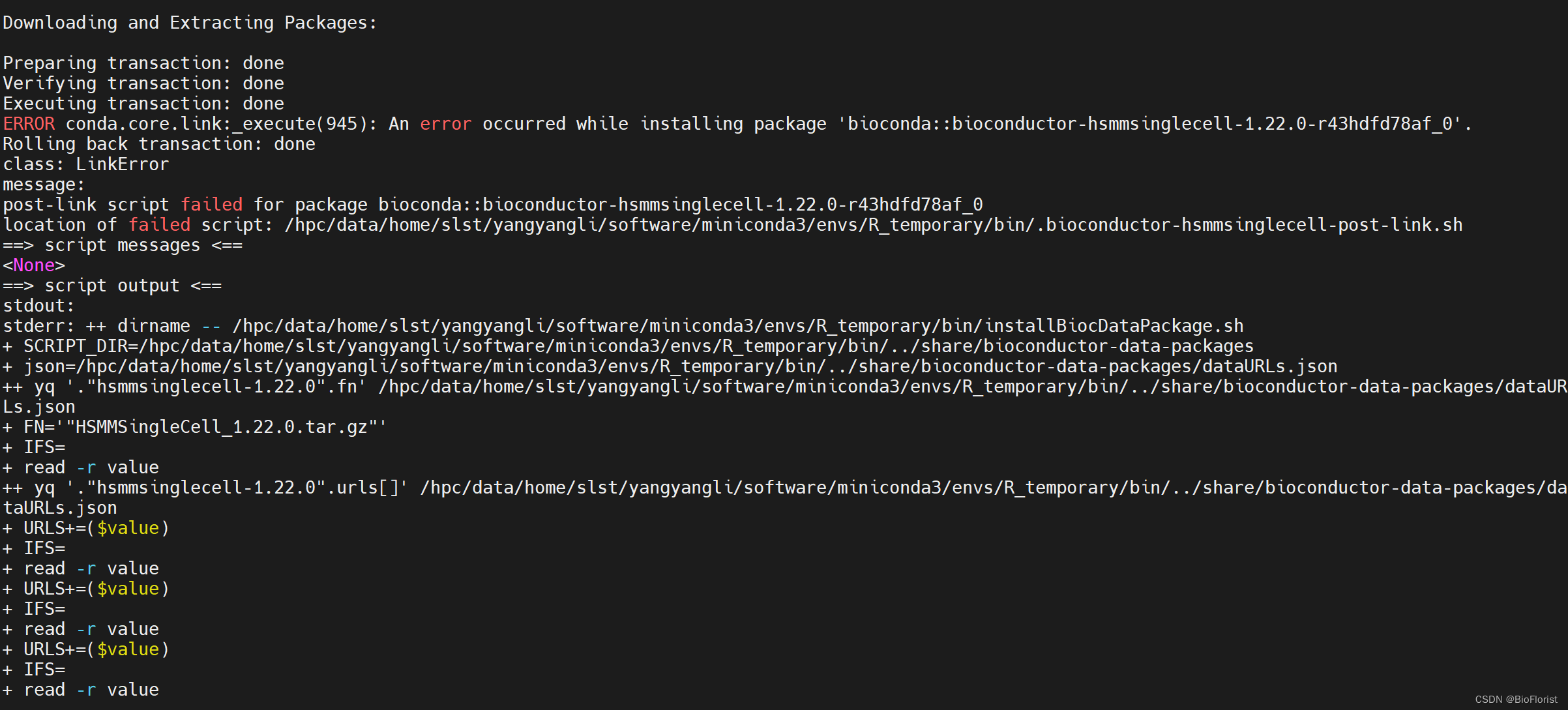
没有sudo权限的情况下好像确实不好装包…
不装了==累了,就写到这。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









