







本文进行光学性质的计算,以ZnO的介电函数为例。Octopus以时间依赖密度泛函理论(TDDFT)为理论基础,对材料施加一定的外界扰动,通过一定时间的传播,搜集相应的响应数据,进而得出各种光学性质。
整个计算同样是分为两步,首先计算基态(gs),再计算时间传播(td)。
基态计算的inp文件如下:
CalculationMode = gs
FromScratch = yes
PeriodicDimensions = 3
ParKPoints = auto
BoxShape = parallelepiped
ExperimentalFeatures=yes
PseudopotentialSet = pseudodojo_pbe # 使用pbe赝势
%Spacing # 单位 Bohr
0.20 | 0.20 | 0.20
%
%LatticeVectors # 相应晶格参数可从 https://materialsproject.org/ 网站上得到
1.0 | 0.0 | 0.0
-0.5 | sqrt(3)/2 | 0.0
0.0 | 0.0 | 1.0
%
a = 3.289*angstrom
b = 5.307*angstrom
%LatticeParameters
a | a | b
%
%ReducedCoordinates
"O" | 1/3 | 2/3 | 0.379214
"O" | 2/3 | 1/3 | 0.879214
"Zn" | 1/3 | 2/3 | 0.000000
"Zn" | 2/3 | 1/3 | 0.500000
%
%KPointsGrid
8 | 8 | 6
%
KPointsUseSymmetries = yes
%SymmetryBreakDir
1 | 0 | 0
%
DFTULevel = dft_u_empirical # 使用DFT+u 来扩展带隙
%Species
"Zn" | species_pseudo | hubbard_l | 2 | hubbard_u | 12.8*eV
"O" | species_pseudo | hubbard_l | 1 | hubbard_u | 5.29*eV
%
# DFT+u 理论参数取自文献: Luis A. Agapito, Stefano Curtarolo, and Marco Buongiorno Nardelli.
#Reformulation of DFT+u as a pseudohybrid hubbard density functional
#for accelerated materials discovery. Phys. Rev. X, 5:011006, Jan 2015.
然后进行时间传播的计算
CalculationMode = td # td计算
FromScratch = yes
PeriodicDimensions = 3
ParKPoints = auto #自动分配k点以进行并行计算
ExperimentalFeatures=yes #使用实验特性
PseudopotentialSet = pseudodojo_pbe #使用pbe赝势
BoxShape = parallelepiped #模拟盒形状为平行正六面体
%Spacing #单位为Bohr
0.2 | 0.2 | 0.2
%
%LatticeVectors
1.0 | 0.0 | 0.0
-0.5 | sqrt(3)/2 | 0.0
0.0 | 0.0 | 1.0
%
a = 3.289*angstrom
b = 5.307*angstrom
%LatticeParameters
a | a | b
%
%ReducedCoordinates #约化坐标
"O" | 1/3 | 2/3 | 0.379214
"O" | 2/3 | 1/3 | 0.879214
"Zn" | 1/3 | 2/3 | 0.000000
"Zn" | 2/3 | 1/3 | 0.500000
%
%KPointsGrid
8 | 8 | 6
%
KPointsUseSymmetries = yes #k点的设置,对称性和对称性破坏方向均需要与gs设置相同
%SymmetryBreakDir
1 | 0 | 0
%
DFTULevel = dft_u_empirical #理论等级修正为DFT+U
%Species
"Zn" | species_pseudo | hubbard_l | 2 | hubbard_u | 12.8*eV
"O" | species_pseudo | hubbard_l | 1 | hubbard_u | 5.29*eV
%
%GaugeVectorField #施加外界扰动电磁场,以速率规范(velocity gauge)的形式给出
1|0|0 #单位为Hartree
%
TDPropagator = aetrs #传播子算法
TDTimeStep = 0.02 #时间步长,单位是 hbar/Hartree, 选择方法在“注1”中
TDPropagationTime = 200 #总传播时间
TDExponentialMethod = lanczos
TDExpOrder = 16
PropagationSpectrumDampMode = polynomial
PropagationSpectrumMaxEnergy = 5*eV
计算结束后,相应信息储存在 ./td.general 文件夹下的 gauge_field 文件中。 Octopus自带小程序来处理这个文件,只需要在含有inp文件的目录下(即 ./ 目录)运行 oct-dielectric-function 命令,小程序自动运行,就会在 ./td.general 文件夹中生成 “dielectric_function”, “inverse_dielectric_function” 和 “chi” 这3个文件。 其中 “dielectric_function” 文件即包含有介电函数的信息。其文件内部结构如下:
# energy Re x Im x Re y Im y Re z Im z
共7列,分别是:能量,介电函数在x方向的实部,虚部,在y方向实部,虚部,z方向实部,虚部。
使用画图小软件gnuplot,简单画图,即可看x方向虚部。
gnuplot
plot "dielectric_function" u 1:3 w l
还可以通过介电函数实部和虚部来计算吸收谱:
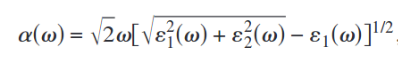
注1:时间步长的确定
时间步长的选择十分重要,太大的步长会导致计算发散,太小的步长又会浪费计算时间。合理的选择思路如下:在不施加外界微扰的情况下进行时间传播计算,传播两百步左右,由于未施加扰动,系统始终处于基态,所以在第一步的总能量和第二百步的总能量应该相同,但是实际计算中,如果时间步长太大,起点和终点的总能量会略有不同,更大的时间步长甚至会导致总能量发散,出现NaN字样,即 Not A Number。但是,起点和终点的总能量调节至完全相同需要很小的步长,会浪费计算时间,所以,选择一个好的步长是十分重要的。














 989
989











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









