







 本文介绍了利用一肽频数、二肽频数、氨基酸亲疏水性倾向及PSSM进行蛋白质二级结构预测,通过支持向量机(SVM)算法构建模型。在不同数据集上,特征组合对模型性能影响显著,其中二肽频数在某些数据集上表现出色,而PSSM特征在所有数据集中都有助于提高预测准确性。
本文介绍了利用一肽频数、二肽频数、氨基酸亲疏水性倾向及PSSM进行蛋白质二级结构预测,通过支持向量机(SVM)算法构建模型。在不同数据集上,特征组合对模型性能影响显著,其中二肽频数在某些数据集上表现出色,而PSSM特征在所有数据集中都有助于提高预测准确性。
简介:
基于蛋白质残基统计信息(一肽频数和二肽频数)、物化属性(亲疏水性倾向)、进化信息(特异位置得分矩阵)进行特征表示,使用支持向量机算法构建蛋白质二级结构预测模型。
一、背景
1、蛋白质
(1)蛋白质组成
蛋白质由氨基酸组成。氨基酸序所包含的基本信息为组成蛋白质的氨基酸种类和氨基酸的排列顺序。如蛋白质S的氨基酸序列可以表示为:
![]()
其中,AA 代表20种氨基酸,Ri 表示蛋白质第i个位置上的氨基酸,L为蛋白质序列长度。
(2)蛋白质结构
蛋白质有四级结构:一级结构、二级结构、三级结构和四级结构。一级结构即为氨基酸序列,二级结构分为α、β、α/β、α+β四种。
蛋白质的一级结构决定了蛋白质的高级结构,而疏水性残基决定了蛋白质二级结构的相对位置。
注:残基为氨基酸序列中的连续片段;
疏水性:每个氨基酸均有疏水性指数和亲水性指数两个物化属性;
2、意义
蛋白质二级结构既可以应用于进一步预测蛋白质的三级结构和四级结构,又能应用于推测蛋白质的功能,因此蛋白质二级结构预测是蛋白质研究的重要组成部分。
3、难点
不同蛋白质的氨基酸序列长度不同,那么如何在保留尽可能多的氨基酸序列信息的同时将其转化为等长的特征数据?
二、特征表示
1、残基统计信息
残基,即为氨基酸序列的连续片段,可以由n(n>1)个氨基酸组成。
注:n=1时称为一肽,n=2时称为二肽,以此类推;
(1)一肽频数
由于氨基酸的种类是固定的,那么可以将每个蛋白质的20种氨基酸出现的频数表示为一个 1x20 维矩阵,即为该蛋白质的一肽频数。比如蛋白质S的氨基酸序列为:
S = A D C F H C E A H H G H
那么其一肽频数(YT)可以表示为:
YT = [ 2 2 1 1 1 1 4 0 0 0 0 0 0 0 0 0 0 0 0 0 ]
(2)二肽频数
一肽频数只考虑到单个氨基酸的频数信息,那么氨基酸与氨基酸之间的结合信息该如何表示呢?
同样的,可以统计蛋白质中任意两种氨基酸连续出现的频数,形成一个 20x20 维矩阵,即为该蛋白质的二肽频数。比如蛋白质S的二肽频数(ET)可以表示为:

2、物化属性
氨基酸有疏水性指数和亲水性指数两个物化属性。
比较氨基酸的亲水性指数和疏水性指数的大小可以判断蛋白质更偏向于亲水性所倾向的二级结构,还是更偏向于疏水性所倾向的二级结构。20种氨基酸的亲疏水性指数如下表所示:
| 氨基酸 |
A |
D |
C |
E |
F |
G |
H |
I |
K |
L |
| 疏水性 |
0.62 |
0.29 |
-0.9 |
-0.74 |
1.19 |
0.48 |
0.4 |
1.38 |
-1.5 |
1.06 |
| 亲水性 |
-0.5 |
-1 |
3 |
3 |
-2.5 |
0 |
-0.5 |
-1.8 |
3 |
-1.8 |
| 氨基酸 |
M |
N |
P |
Q |
R |
S |
T |
V |
W |
Y |
| 疏水性 |
0.64 |
-0.78 |
0.12 |
-0.85 |
-2.53 |
-0.18 |
-0.05 |
1.08 |
0.81 |
0.26 |
| 亲水性 |
-1.3 |
0.2 |
0 |
0.2 |
3 |
0.3 |
-0.4 |
-1.5 |
-3.4 |
-2.3 |
注:疏水性来自JACS, 1962, 84: 4240-4246. (C. Tanford);
亲水性来自PNAS, 1981, 78:3824-3828 .(T.P.Hopp & K.R.Woods);
根据蛋白质S的氨基酸序列,该蛋白质的亲水性指数和疏水性指数为:

那么结合蛋白质S的一肽频数(YT),将蛋白质S的亲疏水性倾向(Hydr)表示为:
![]()
3、进化信息
特异位置得分矩阵(简称PSSM),是一种基于蛋白质进化信息的打分矩阵,表示20种氨基酸在蛋白质中特定位置出现的概率。PSSM得分普遍表现为正或负整数,正分数表明给定的氨基酸替代发生的比预期的要频繁,负分数表明替代发生的比预期的要少。大的正分数通常表示关键的功能残基,普遍这可能是其他分子间相互作用所需的活性位点残基或残基。PSSM可以从PSI-BLAST(https://www.ebi.ac.uk)获得。由于PSSM是一个 L×20 维的矩阵(L为基酸序列长度),本文采用矩阵线性转化的方法,将PSSM转化为矩阵P具体如下:
![]()
矩阵P是20×20维矩阵,且P是对称矩阵,为了减少冗余的特征属性和计算,本文只采用P的下三角矩阵,并将其转化为一个1×210维矩阵PSSMfeature。

三、模型构建
1、数据
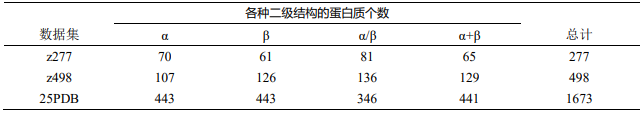
数据分为 3 个数据集,其中数据集 z277 和数据集 z498 的蛋白质相似度较高,数据集 25PDB 与其他两个数据集的蛋白质相似度较低。
注:数据集的蛋白质相似度高,即表示数据集内不同蛋白质的相似度高;
2、算法
数据集样本量较少,本文采用svc算法来训练模型。
(1)多分类问题
SVC主要针对二分类问题,目前将svc使用于多分类问题有2种解决方法。
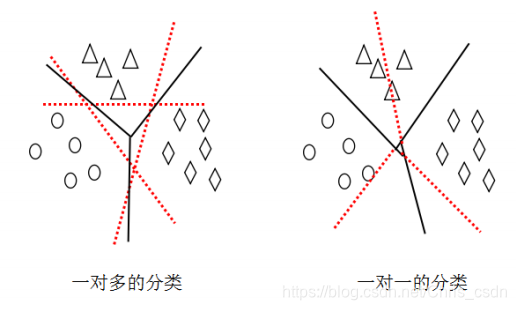
1)一对多的分类
构造Q(Q为类别的个数)个二分类svc模型,每个模型划分一类样本。
比如第i(1≤i≤Q)个模型负责划分第m(1≤m≤Q)类样本,那么模型的结果只有两种:样本预测为第m类和样本预测为不是第m类。
2)一对一的分类
构造Q(Q-1)/2(Q为类别的个数)个二分类svc模型,每个模型划分任意两类样本。也就是说每一类样本点都要与剩下的Q-1类样本点一一进行比较。比如第i(1≤i≤Q(Q-1)/2)个模型负责划分第m(1≤m≤Q)类和第n(1≤n≤Q)类样本,那么模型的结果只有两种:样本预测为第m类和样本预测为第n类。
由于一对一的分类方法思路较清晰且保留了不同类之间的分割超平面,因此采用一对一的分类方法。
(2)核函数
1)线性核函数:主要用于线性可分的情况,参数少速度快。
2)多项式核函数:全局核函数,因此容易过拟合。
3)高斯核函数:局部核函数,对存在噪声的数据具有很好的抗干扰能力。
本文的数据集特征维度较高(20-610维),核函数选用linear kernel。
3、模型训练
(1)标准化
为了使不同的特征属性具有可比性,且提高模型的分类效果,对特征进行标准化。
(2)留一交叉验证
数据集样本量较小,训练集与测试集有限,采用留一交叉验证可较客观地评估模型的效果。
(3)模型评价
采用acc和mcc指标来评价模型的效果。acc和mcc指标的值越大,分类模型的效果越好。
|
|
预测类别 |
||
| Yes |
No |
||
|
实际分类 |
Yes |
TP |
FN |
| No |
FP |
TN |
|
![]()
![]()
4、模型效果
(1)数据集 z277
| 特征 |
acc (%) |
||||
| α |
β |
α/β |
α+β |
all |
|
| 一肽频数 |
84.838 |
87.004 |
85.560 |
82.671 |
70.036 |
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 4189
4189

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









