







目录
-
软件下载
-
软件介绍
-
安装教程
-
使用教程
软件下载
[软件名称]:Materials Studio 2020
[界面语言]:英文
[安装环境]:Win11/Win10
[系统位数]:32位&64位
[软件类型]:分子材料计算模拟软件
[下载链接]:宫粽号回复【Materials Studio】获取完整安装教程及使用教程
解压密码在宫中号【元素魔方科研服务】获取 https://pan.baidu.com/s/1cxYcIjzQVTPvrdydQ8M8QA?pwd=1234
https://pan.baidu.com/s/1cxYcIjzQVTPvrdydQ8M8QA?pwd=1234
软件介绍
Materials Studio软件(简称MS)是一款可以快速入门DFT计算的量化软件,它集建模与计算为一体,可以非常方便地进行计算。以我们专业需求为例,它可以计算材料的晶体性质(电子、光子、弹性、振动与热力学性质等)、表面性质(表面能、静电势、功函数等)、表面吸附性质(吸附能、差分电荷密度等)及过渡态搜索等性质。该软件具有windows与linux版本,对没有编程基础的同学非常友好。
安装教程
注意事项
①解压安装包和软件安装时,关闭杀毒软件,否则安装文件容易被误删除;
②安装软件时,要右击压缩包,把压缩包解压出来,不要直接双击压缩包;
③安装包及软件安装路径,最好是英文或数字,如有中文路径,容易报错;
1.安装包大小为 1.86G,安装之前关闭防火墙和杀毒软件
2.解压安装包之后,以管理员身份运行 setup 程序
3.点击下一步
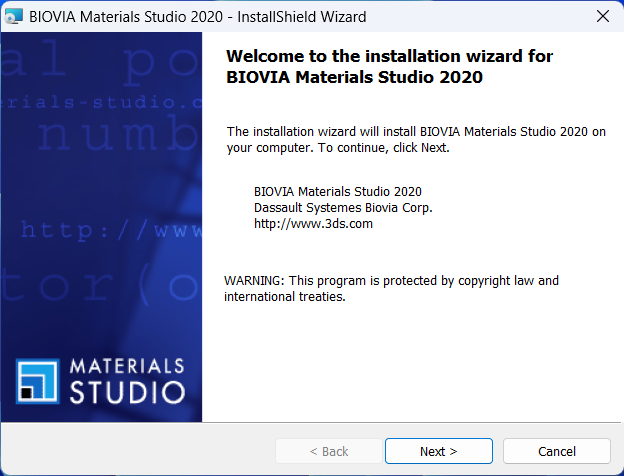
4.这两行随意输入内容,点击下一步
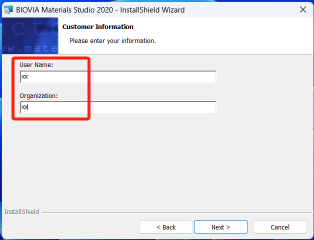
5.设置安装路径,最好只改首字母的盘符,路径中不要有中文,点击下一步
6.点击下一步
7.点击下一步
8.点击安装
9.安装过程会自动弹窗,耐心等待一会
10.安装结束,取消勾选,点击完成
11.在开始菜单找到 Materials Studio 2020 ,把它拖到桌面
12.选中快捷方式右键打开文件夹所在位置,把安装包里 Crack 文件夹中的 msi2020.lic 复制到刚刚快捷方式打开的文件路径下
13.以记事本格式打开 msi2020.lic,界面如下
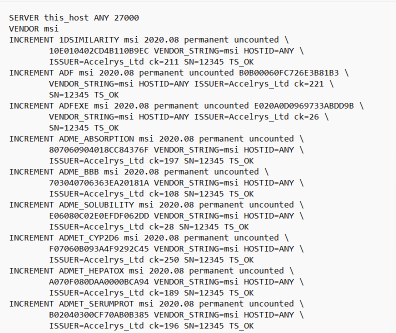
14.选中此电脑右击属性,复制设备名称
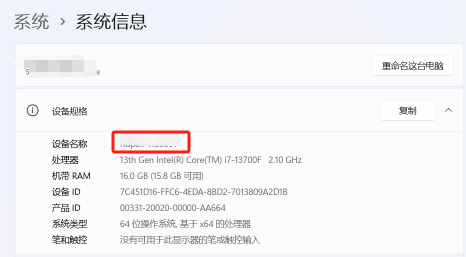
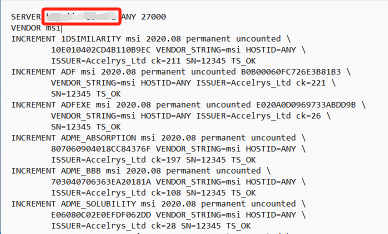
15.回到记事本文件,将第一行的 this_host 替换为计算机全名,然后保存关闭(如果这一步保存遇到问题,可以使用 Notepad++ 去修改)
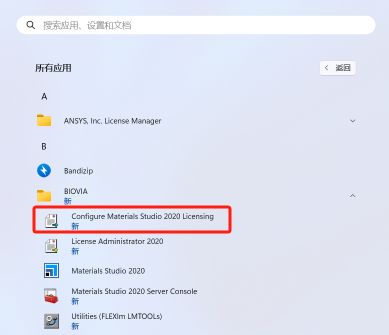
16.在开始菜单找到配置程序,右键选择以管理员身份运行
17.勾选最后一个,点击下一步
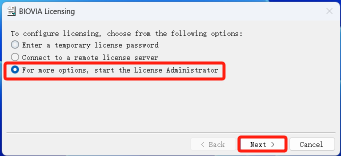
18.点击证书文件下的安装证书,点击浏览
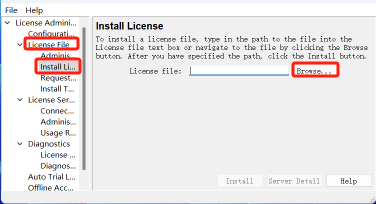
19.打开安装目录,找到刚刚修改的 msi2020.lic 然后点击打开
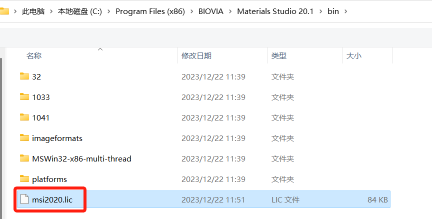
如果不容易找到,可以将下方的所有文件改为证书文件
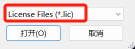
20.点击安装
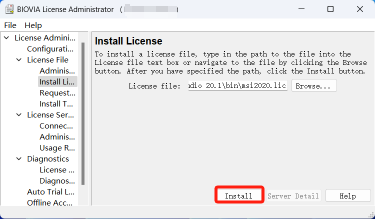
21.点击 OK
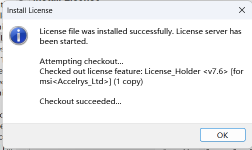
22.打开软件,界面如下
使用教程
在学习软件前,我们要明白软件仅是一个工具,最重要的还是需要知道我们想要计算什么。比如我想分析一个小分子吸附到固体表面的吸附能是多少,那么我们需要收集以下信息:
1. 小分子的分子式,如CO,H2O或其他;
2. 固体材料结构,包括这个固体是什么元素构成,它的晶体结构是什么(比如面心立方、体心立方等)或者需要知道它的空间群信息;
3. 小分子吸附到固体材料结构时,是小分子的哪个原子与固体表面的哪种元素相结合。
收集到以上信息以后,我们便可以进入软件进行操作。首先需要了解软件的基本操作界面。
【MS基本操作界面】
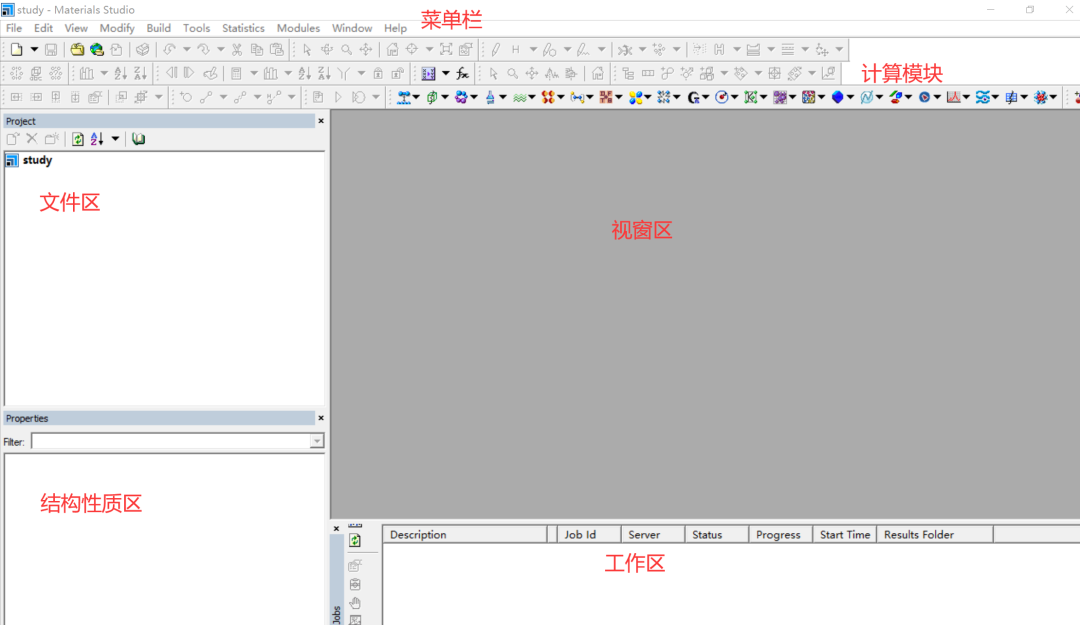
首先启动MS软件,进入如下界面,界面主要分为六个部分,分别是:工程区(文件目录)、视窗区、性质区、工作区、菜单栏以及计算模块。
【建立新文件】
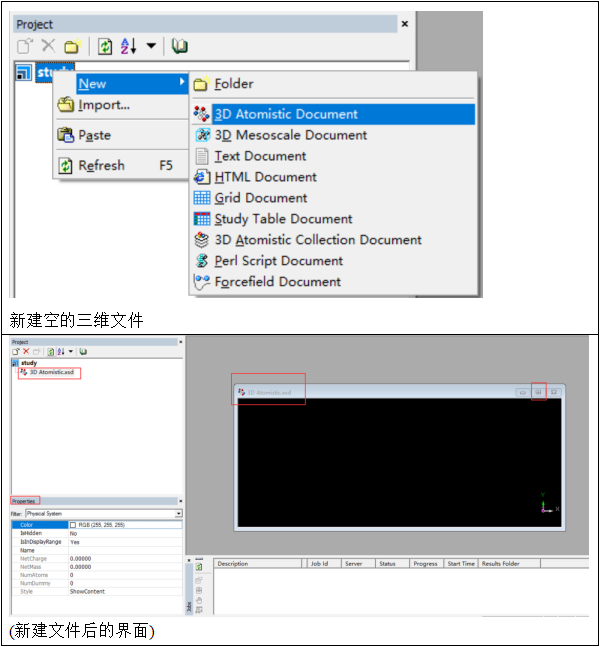
在文件区域中,我们可以添加或删除文件,操作方式与日常电脑上的文件管理类似。举例来说,若需添加一个新的空白3D文件,首先点击文件名,右键选择‘new’,然后选择‘3D Atomistic Document’,即可创建出新的文件(见下图)。
下面我们以计算CO在Pt(111)晶面上的吸附能为例,继续进行操作。主要需要分为以下几个步骤:
(1) 将金属Pt的结构文件导入
(2) 接着对该结构沿着(111)晶面方向进行切面
(3) 设置好材料大小及层数
(4) 进行结构优化,计算该材料的能量,得到E1
(5) 接着使用“小铅笔”工具,在该材料表面进行CO的吸附
(6) 进行结构优化,得到该状态下的能量E2
(7) 此外新建文件,画一个CO分子,进行结构优化并计算CO气态分子的能量,得到E3
(8) 计算吸附能
【导入结构文件】
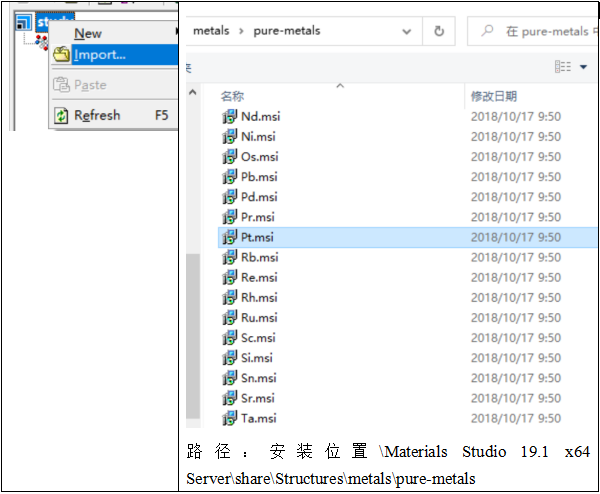
MS自带有材料数据库,位置在安装文件位置+Materials Studio19.1x64Server+share+Structures,根据材料基本分类就可以找到相应的结构文件,如果数据库没有,可以在下面网站找一下。
[materialsproject.org,https://www.ccdc.cam.ac.uk/structures,http://nanocrystallography.org]
下面从MS自带数据库中导入金属Pt的结构文件
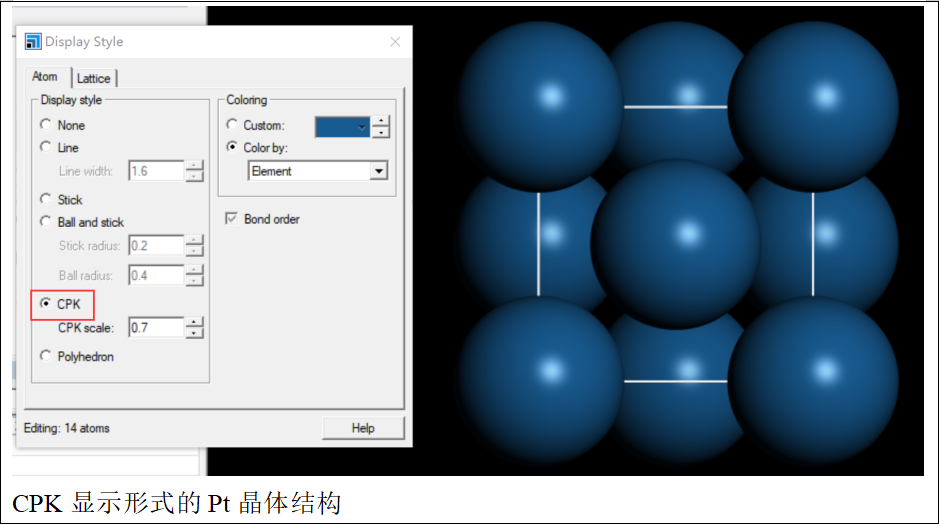
接下来,将鼠标放在视窗窗口上,右键选择“display style”,然后选择显示模式为“CPK”,这样就能更清晰地观察晶体结构。
【切面】
下面进行切面操作,在菜单栏选择“bulid”-“surfaces”-“cleave surface”
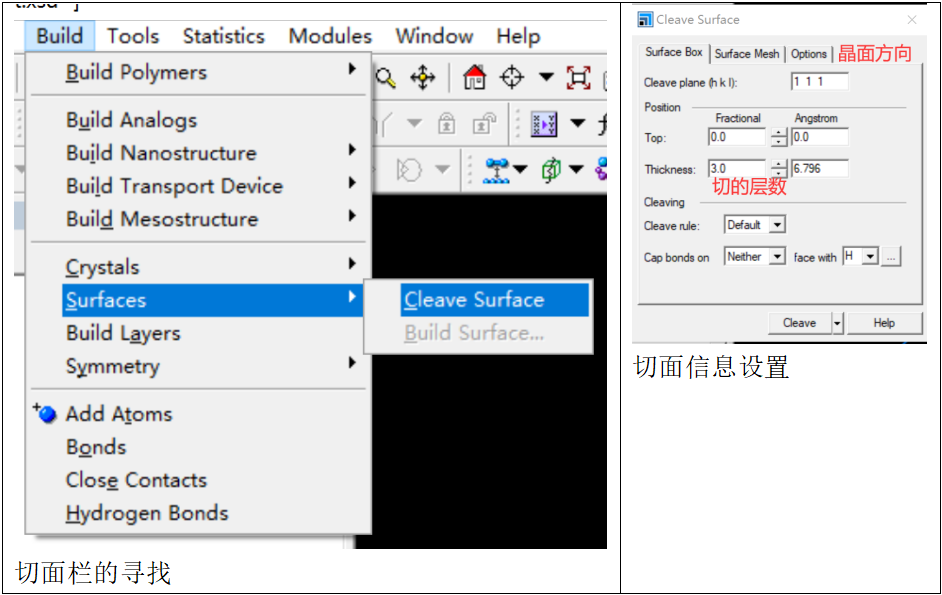
设置切面信息(包括期望的晶面方向和切割的层数),得到三层的Pt(111)材料;但显示效果不太明显,因此将鼠标放在视窗窗口,调出“display style”,在lattice参数中,将u和v均增加到4,即可得到(4×4)的三层Pt(111)材料。
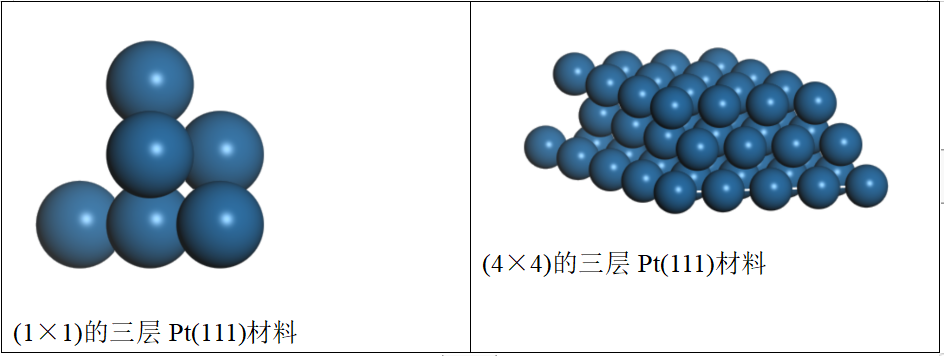
【结构优化】
在对该材料进行结构优化前,需要先建立一个真空层,在菜单栏中选择:
“build”-“crystals”-“build vacuum slab”,设置真空层厚度为25埃(15~30均可)
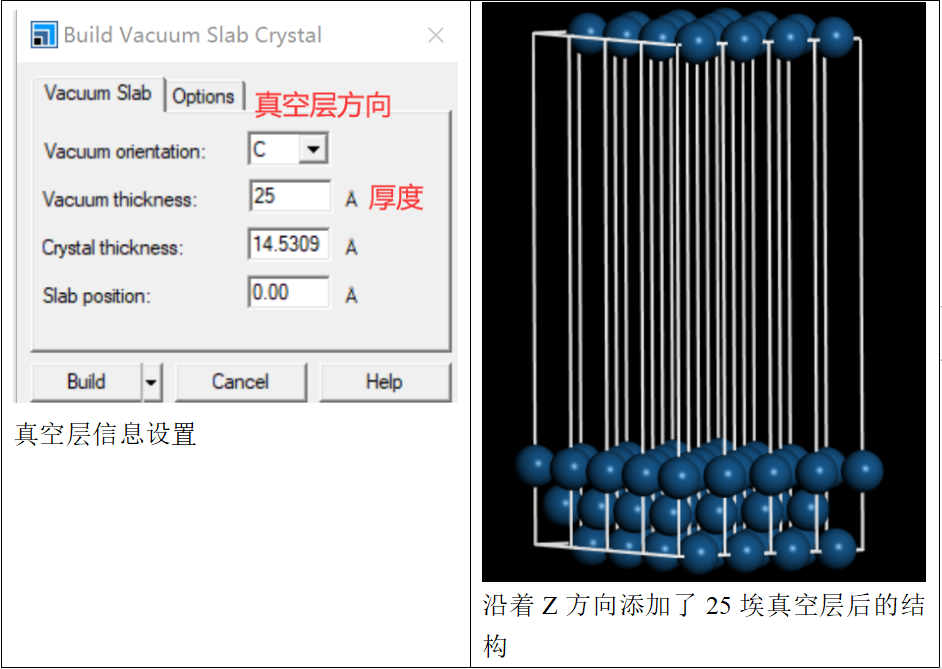
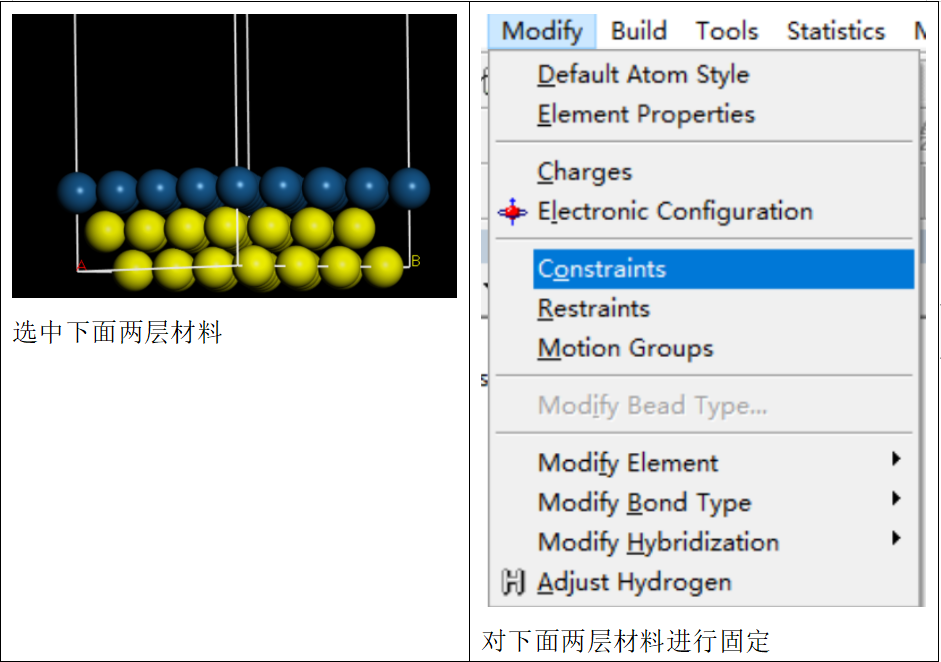
接着将材料结构设为超胞形式:
“build”-“symmetry”-“supercell”。并将材料的第一第二层材料进行固定(选中第一第二层,在菜单栏点击“modify”-“constrains”),以代表体相所处的状态。
接下来进行结构优化以得到稳定的结构,这里选择dmol3模块进行优化。在计算模块中,选择dmol3-caculation,调出计算任务窗口,将计算任务设置为“Geometry optimization”,然后根据文献或学习选择合适的计算条件(如泛函、基组等信息设置),点击“run”即可开始计算。
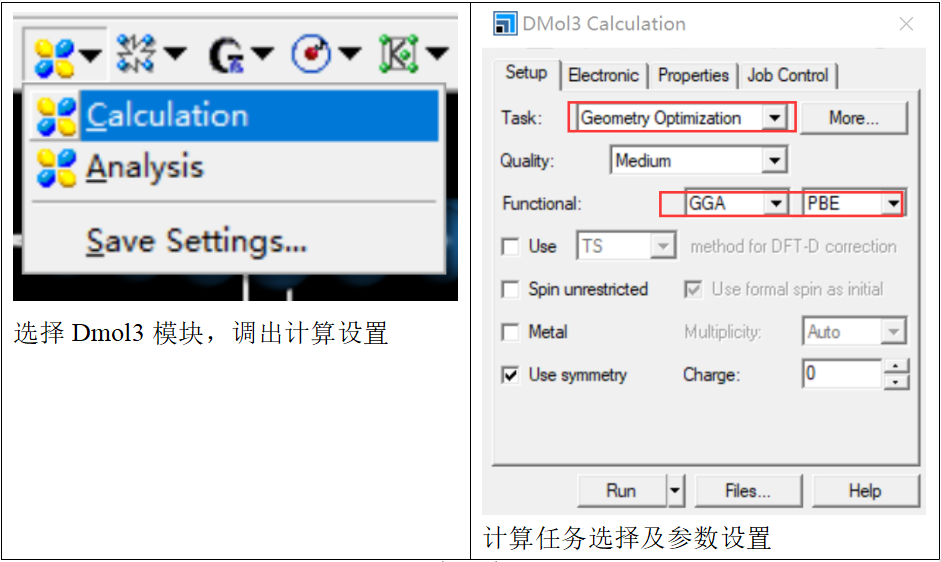
【CO吸附与结构优化】
在已经结构优化完成的(4×4)三层Pt(111)材料上进行CO表面吸附,主要使用“sketch atom”小铅笔在材料表面构建CO。选中CO分子后,可以对其位置进行调整。
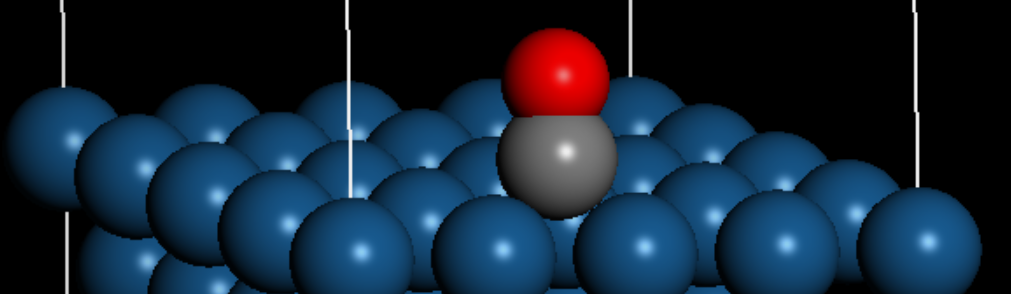
同样地,在dmol3模块中调出计算设置,对该结构进行优化以计算该状态下的能量。此外,需要在一个新的空白3D文件中,同样使用“sketch atom”小铅笔绘制一个CO分子,对其进行结构优化并计算其能量。
通过查阅文献,可以计算出CO在Pt(111)晶面的吸附能。
ps:任何计算都是源于我们的需求,因此在计算前,我们需要明确的知道我们想要计算什么。接着便是查阅资料收集所需要的计算信息,比如前面所提到的晶体结构信息等。


















 964
964

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











