







软件名称 | 主要功能 |
XRD数据分析工具 | |
XPS数据分析工具 | |
XRD数据分析 | |
拉曼光谱分析 | |
核磁共振NMR数据处理 | |
科研图像处理工具 | |
XPS数据分析 | |
观察生物分子微观结构 | |
纳米颗粒粒度分析软件 | |
原子水平晶体结构可视化 | |
文献管理软件 | |
数据分析工具 | |
三维建模和渲染软件 | |
电镜图像分析软件 | |
晶体分子结构图绘制软件 | |
非线性拟合软件 | |
晶体结构解析软件 | |
热化学分析软件 | |
红外数据分析软件 | |
科研绘图软件 | |
分子材料计算模拟软件 | |
XPS和AES数据分析软件 | |
Xcalibur | 质谱数据分析软件 |
Mercury | 观察晶体结构软件 |
Nanoscope Analysis | AFM数据处理软件 |
Image Pro Plus | 图像分析软件 |
ZSimpWin | 阻抗谱拟合软件 |
Masshunter | 质谱采集分析软件 |
Cytoscape | 生物信息分析软件 |
CrysTBox | TEM数据分析软件 |
Demeter | 同步辐射XAFS软件 |
TA advantage | 热重分析软件 |
目录
软件下载
软件介绍
安装教程
使用教程
软件下载
[软件名称]:Gaussian16
[安装环境]:Win11/Win10/Win9/Win8/Win7
[系统位数]:64位
[软件类型]:量子化学计算软件
[下载链接]:公众号回复【Gaussian】
软件资源链接及软件解压密码
关注下方公众号

回复【Gaussian】获取
(ps: 复制括号内红色的关键词回复,不要手动输入,如空格或字母大小写会影响关键词的软件推送)
软件介绍
高斯程序(Gaussian)是做半经验计算和从头计算使用最广泛的量子化学软件,可以研究:分子能量和结构,过渡态的能量和结构,化学键以及反应能量,分子轨道,偶极矩和多极矩,原子电荷和电势,振动频率,红外和拉曼光谱,NMR,极化率和超极化率,热力学性质,反应路径。
服务器、理论计算服务推荐:
我们公司提供高性能计算解决方案,专为使用Gaussian等量子化学软件的用户优化。通过我们的高性能CPU、GPU服务器和集群,您将体验到更快的计算速度。我们的服务适用于人工智能、生信分析、分子模拟、有限元分析、流体力学分析等多个领域。
此外,我们拥有985专业硕博计算团队,经验丰富,能够针对不同专业和领域进行课题建模及模拟计算,为您的每一个数据负责。我们擅长人工智能、分子模拟、量子化学、第一性原理、有限元、CFD流体力学仿真等领域。
点击二维码详细了解相关服务

安装教程
注意事项
①解压安装包和软件安装时,关闭杀毒软件,否则安装文件容易被误删除;
②安装软件时,要右击压缩包,把压缩包解压出来,不要直接双击压缩包;
③安装包及软件安装路径,最好是英文或数字,如有中文路径,容易报错;
1.下载压缩包,并解压到压缩文件
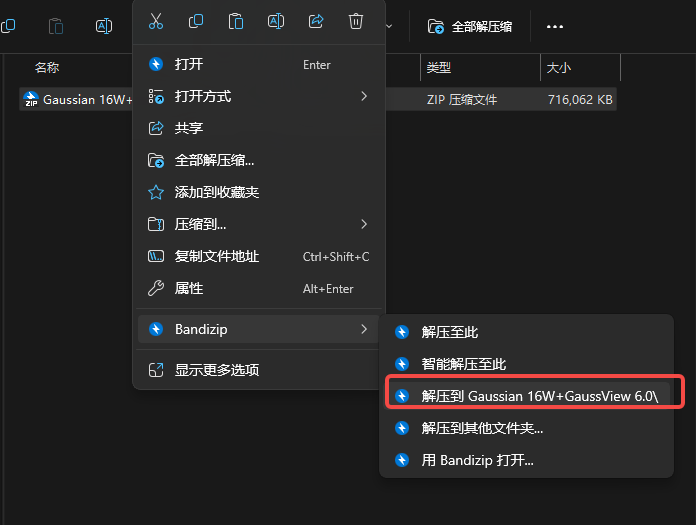
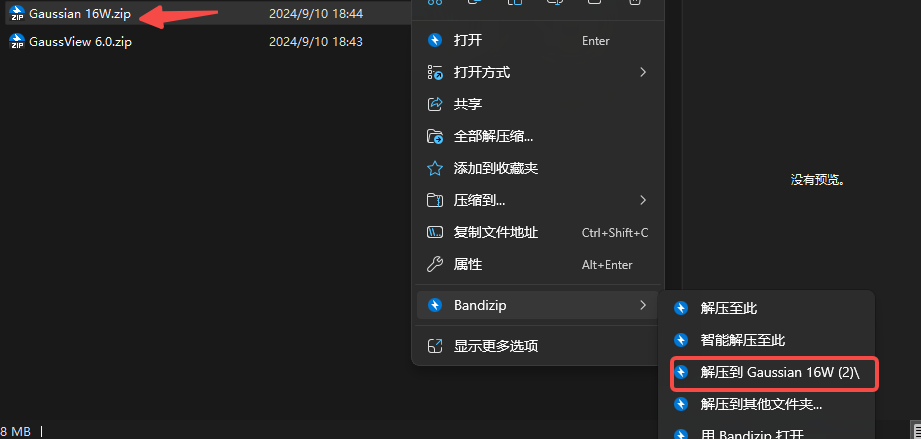
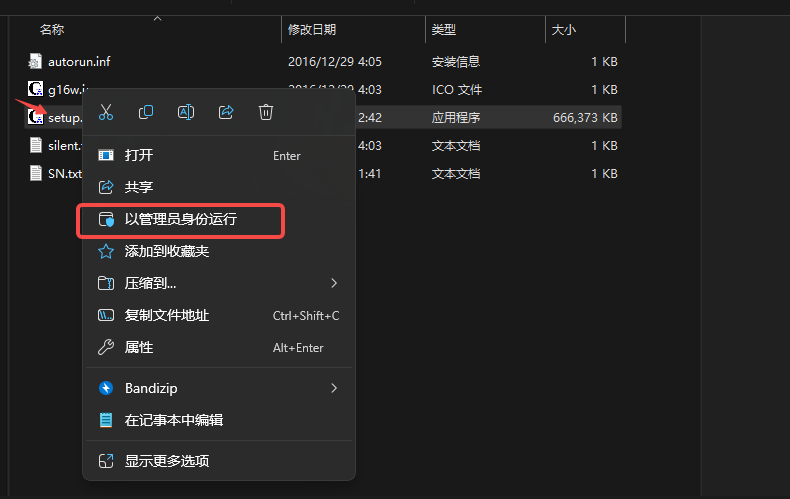
2.右击,以管理员身份运行
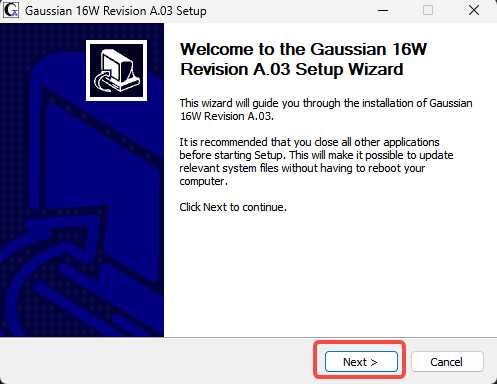
3.Next
4.随意填写用Name和Company,
Serial填写G64294554961898W-1271N,Next
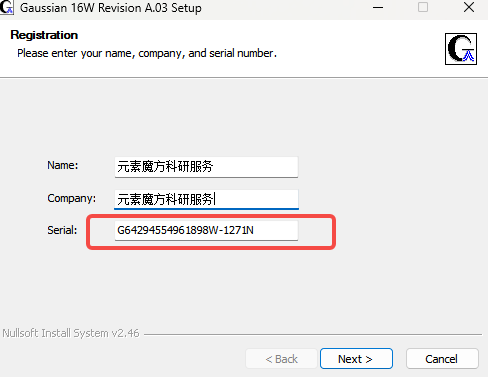
5.确定
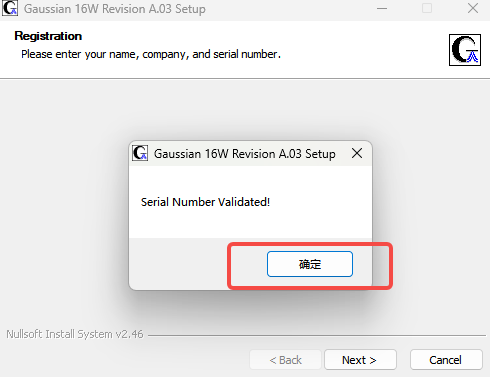
6.Next
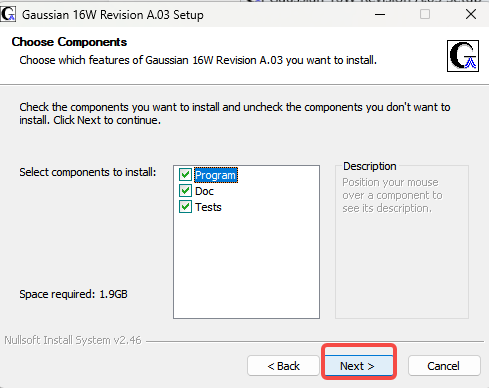
7.更改安装位置(需要的改)
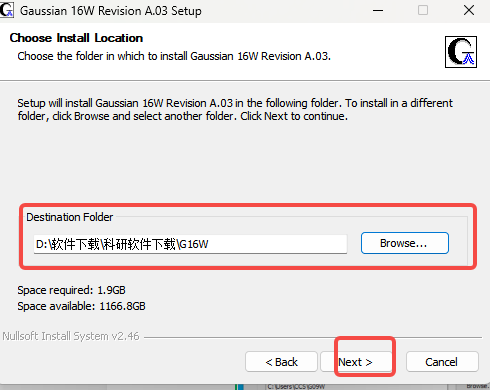
8.Install
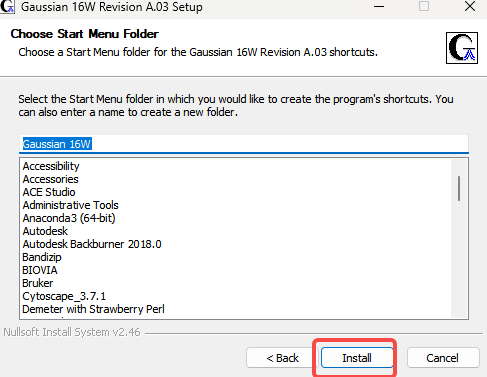
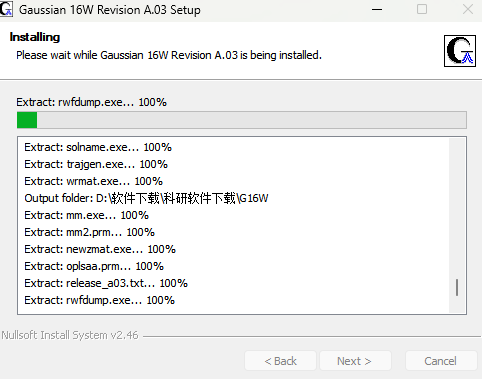
9.创建一个软件缓存位置(建议全英文),确定
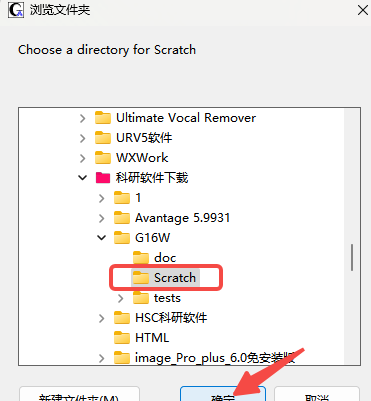
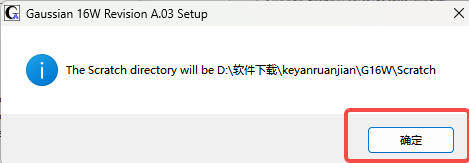
10.确定
11.Finish
12.打开桌面软件,完成
经过上述安装步骤,你应该已经成功安装了 Gaussian 16 。首次启动系统后,你可以根据个人需求进行一些基本配置,以确保软件的最佳性能。
1. 配置 Gaussian 16 的运行环境:配置默认内存和处理器核心数。
2. 准备输入文件:Gaussian 16 的输入文件以 .gjf 为扩展名,包含分子结构、计算方法和其他控制参数。您可以通过文本编辑器(如 Notepad++)手动编辑输入文件。
3. 提交计算任务:提交计算任务后,可以通过查看输出文件实时监控计算进度。
4. Windows版本计算是有上限的,内存上限4G。重度使用建议使用Linux版。
使用教程


以下来源:Fluorescence WORLD公众号
安装软件完毕,打开程序如图3、图4所示。由于篇幅所限,只对其中最简单部分进行介绍。
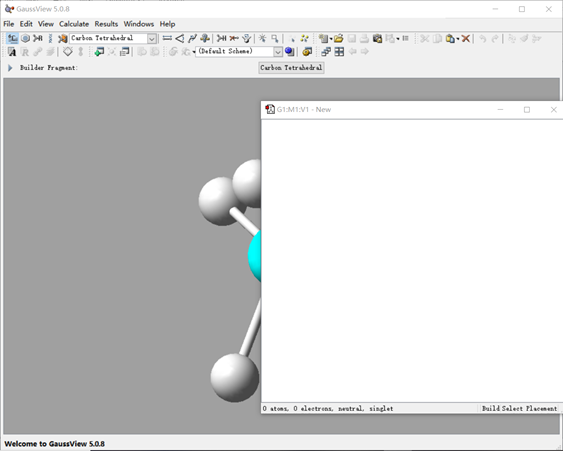
图3 GaussView 5.0的初始界面
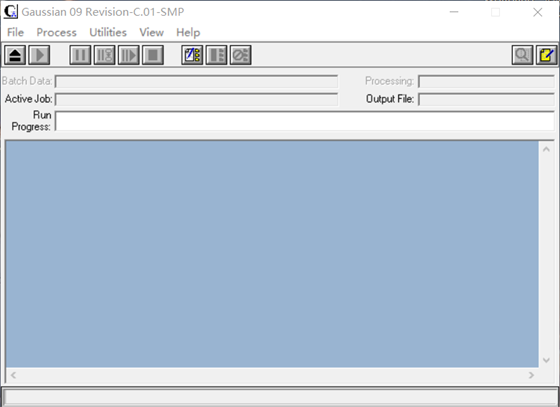
图4 Gaussian09W的初始界面
图3是GaussView5.0的界面,GaussView是为了便于对Gaussian输入文件的坐标输入和部分计算结果的可视化而开发出的衍生软件。该软件可以对物质结构(原子、成键方式、键长、键角、二面角等)进行描绘,并可以直接递交作业于Gaussian(如果已经安装)。分步说明如下:
(1)采用该软件画好物质结构;
(2)点击Calculation-----Gaussian Calculation Set up….,会弹出以下对话框(如图5所示);
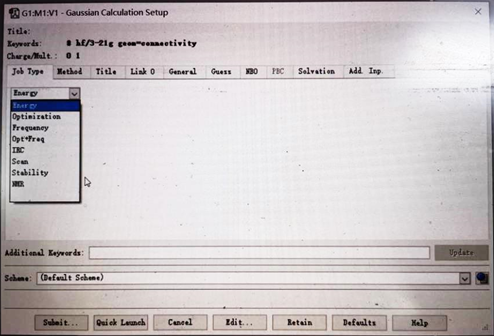 图5 点击Gaussian Calculation Set up…. 后弹出的界面
图5 点击Gaussian Calculation Set up…. 后弹出的界面
(3)JobType(任务类型)下面选择想要进行的任务类型,分别是单点能、结构优化、频率分析、几何优化+频率分析等;
(4)点击Method(方法),选择计算方法以及基组,在Hartree-Fork那一栏,可以选择计算方法(HF,DFT,MP2,CCSD等……),在3-21G那一栏可以选择基组(sto-3G,6-31G,6-311G等……),一般规律是从上到下基组从小变大,一般来说,大基组意味着计算精度更高,也更贵(指耗时更长)。后面的*代表极化函数(在6.0版本中,不再使用*,不过**其实是等效于(d,p));+代表弥散函数,弥散函数一般只在阴离子体系和有弱相互作用情况下加。
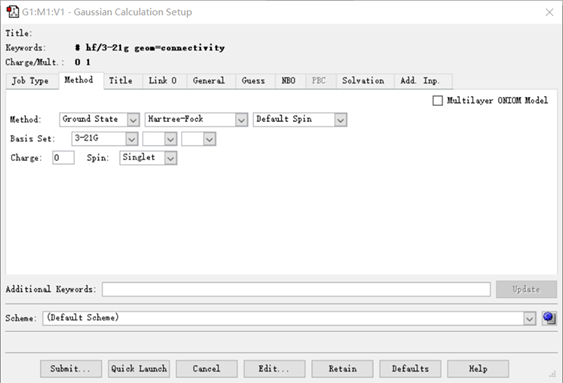 图6 Method下的界面
图6 Method下的界面
(5)还可以在Link 0---options---memory limit 选择给程序使用的内存大小(在6.0版本,还可以指定核数等)。
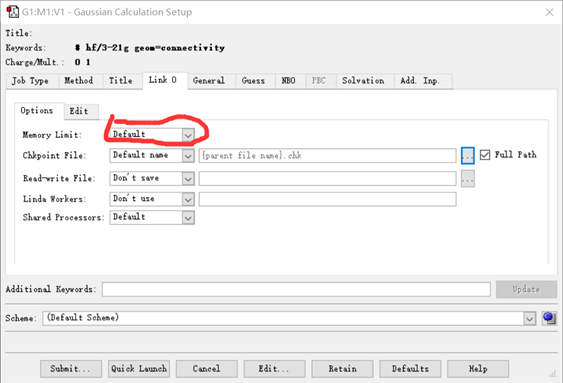 图 7 Link 0下的界面
图 7 Link 0下的界面
(6)确认无误后,点击submit ,会弹出让你保存输入文件(*.gjf)的提示,保存完毕,然后就把作业递交到Gaussian进行计算,最后会得到*.chk和*.log两个文件。可以用写字板等格式打开,找到想要查看的结果;如果进行的任务是结构优化或者频率分析等,也可以用GaussView打开*.chk文件,直接观察分子结构或从Results查看振动分析…。
Gaussian的使用步骤如下:
(1)打开File下的New,会弹出Job Entry 界面(如图8所示)。这里我们需要知道每一栏需要填些什么:%Section确定执行文件名,行首需以%开始,段后无空行,例如:%chk=D:\text\1.chk(有时可不填);RouteSection:行首以#开始,再加指定计算方法、基组、计算项目,段后加空行,例如:# optb3lyp/6-31g(d,p)(这是必须要写出的);Title Section:题目(可以不写);Charge&Multiple:电荷加空格加自旋多重度(正常的分子一般为1),例如:0 1 ;MolecularSpecification:写出分子中各原子坐标(猜测的,未必是完全准确的。例如,笔者某次构建的苯的分子坐标如下:(
C -0.44802866 -0.01792115 0.00000000
C 0.94713134 -0.01792115 0.00000000
C 1.64466934 1.18982985 0.00000000
C 0.94701534 2.39833885 -0.00119900
C -0.44780966 2.39826085 -0.00167800
C -1.14541066 1.19005485 -0.00068200
H -0.99778766 -0.97023815 0.00045000
H 1.49663934 -0.97043415 0.00131500
H 2.74434934 1.18990985 0.00063400
H 1.49721534 3.35048185 -0.00125800
H -0.99793166 3.35054185 -0.00263100
H -2.24501466 1.19023785 -0.00086200
),写入时段后一般要加两空行(现在这一部分常采用从GaussView中画出初猜结构,然后把坐标读取出来,最后把坐标复制过来这一做法,不过空行最好还是加上)
(2)填好后,点击File---save job/ save job as…保存输入文件
之后点击与%Section平行的第一个按钮(即有播放箭头上面且写着“RUN”),
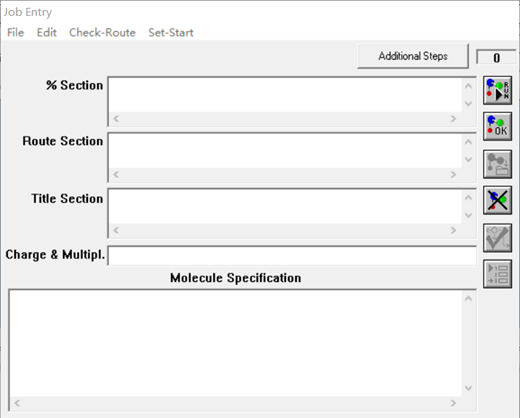
图 8Job Entry 界面
(3)这时会弹出一个保存文件的框,可以自己选择保存位置以及输入保存文件名*.out(这与GaussView不同)。设置完毕,程序开始进行计算。
(4)如果输入正确且输入较为合理,程序界面最后会得到如图9红色部分标记的一句话,说明程序正常结束。
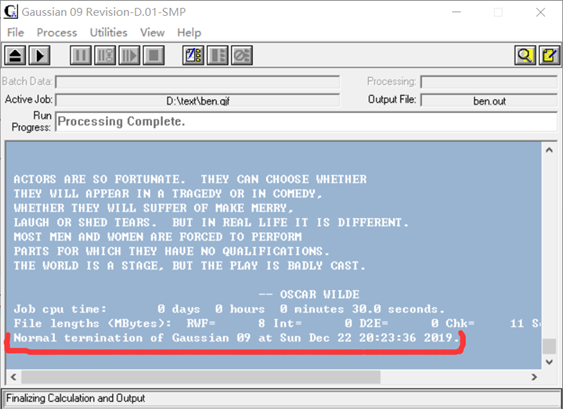
图 9正常结束标记(红色标记部分)
(5)可以直接用写字板等直接打开*.out文件,查看输出结果;也可以用GaussView打开它,查看几何结构和其他信息,上文已有叙述。
特别说明:本文只是一个简单的介绍,实际操作中,常常会出现各种各样的BUG,如何解决这些BUG,也是使用软件的重中之重。
温馨提示:分享的所有软件,均由互联网中的资源整理所得,仅限学习交流,切勿商用,侵删!
—The end—
更多资源请关注元素魔方科研服务

















 1053
1053

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









