







论文简介

标题:Gallium cluster-promoted In2O3 catalyst for CO2 hydrogenation to methanol
作者:Yuxiang Yang, Linlin Wu, Bingqing Yao, Lei Zhang, Munam Jung, Qian He, Ning Yan, and Chang-Jun Liu
期刊:ACS Catal.(IF:12.9)
原文链接:https://doi.org/10.1021/acscatal.4c03045
研究背景
这篇论文的研究背景基于二氧化碳(CO2)加氢转化为甲醇这一过程的广泛关注。随着低碳燃料需求的增加,学术界和工业界对CO2氢化制甲醇的兴趣不断上升。传统的甲醇合成催化剂主要基于铜,但这些催化剂在使用CO2作为碳源时面临选择性低和快速失活等问题。因此,越来越多的研究集中在提高催化剂性能,尤其是非铜基催化剂的开发。
氧化铟(In2O3)催化剂由于其优越的甲醇选择性而受到关注,这种高选择性主要归因于其表面氧空位。然而,氧化铟催化剂在CO2转化率和稳定性方面仍存在不足。为了解决这些问题,研究者们尝试通过添加金属助剂(例如钯、铑、铜等)来增强催化剂的活性。特别地,镓(Ga)被证明在铜基催化剂中能够促进甲醇生产,因此,本研究首次将镓引入In2O3催化剂中,旨在探索镓对CO2氢化反应的促进作用。
论文速览
这篇论文的研究旨在探讨掺镓的氧化铟催化剂在二氧化碳加氢制甲醇反应中的表现。传统的铜基催化剂在该反应中存在选择性和稳定性问题,氧化铟(In2O3)因其高选择性而成为研究的重点,但其在转化率和长期稳定性方面仍有待改善。为了提高催化性能,研究者首次引入镓作为助剂,旨在增强In2O3催化剂的性能。
在研究方法上,论文通过催化剂的合成、表征及催化性能测试,评估了不同镓负载量对In2O3催化剂性能的影响。实验采用了X射线光电子能谱(XPS)、高分辨透射电子显微镜(HR-TEM)等手段,对催化剂的结构和表面性质进行表征,并通过CO2加氢实验,测定其甲醇选择性、CO2转化率和时空收率(STY)。此外,论文使用了密度泛函理论(DFT)计算,进一步阐明了反应机理。
研究结果表明,掺入少量镓显著提高了In2O3催化剂的活性和选择性,特别是在较低的反应温度下表现出优异的甲醇生成能力。相比未掺杂的氧化铟催化剂,掺镓催化剂表现出更高的CO2转化率和更好的长期稳定性。这一研究为开发高效的CO2氢化制甲醇催化剂提供了新的思路和参考。
图文导读

图1. CO2加氢反应性能。(A)CO2转化率和甲醇选择性,(B)1.0%Ga/In2O3、5.0%Ga/In2O3和原始In2O3催化剂的甲醇时空收率(STY),(C)1.0%Ga/In2O3催化剂的长期稳定性。反应条件:5 MPa,300°C,气体小时空速(GHSV)= 21,000 cm³·h⁻¹·gcat⁻¹,H2/CO2 = 4/1。
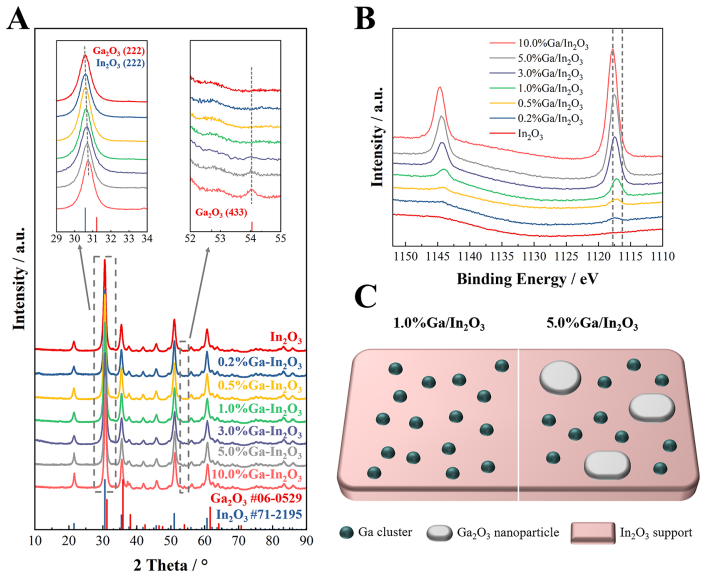
图2. 催化剂结构。(A)原始In2O3和Ga/In2O3催化剂的X射线衍射(XRD)图谱,(B)原始In2O3和Ga/In2O3催化剂的Ga 2p X射线光电子能谱(XPS),(C)1.0%和5.0%Ga/In2O3催化剂的结构示意图。
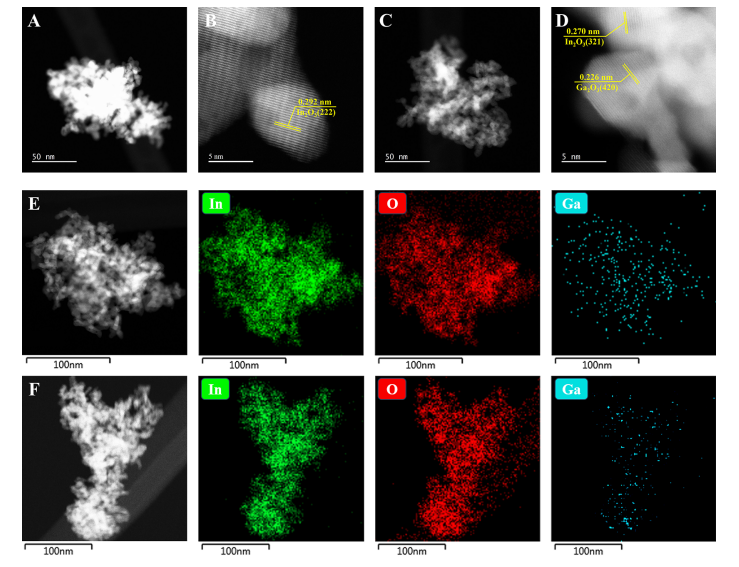
图3. 催化剂形貌。(A, B)1.0%Ga/In2O3催化剂的高角环形暗场扫描透射电子显微镜(HAADF-STEM)图像,(C, D)5.0%Ga/In2O3催化剂的HAADF-STEM图像,(E)1.0%Ga/In2O3和(F)5.0%Ga/In2O3催化剂的能量色散X射线(EDX)元素分布图。
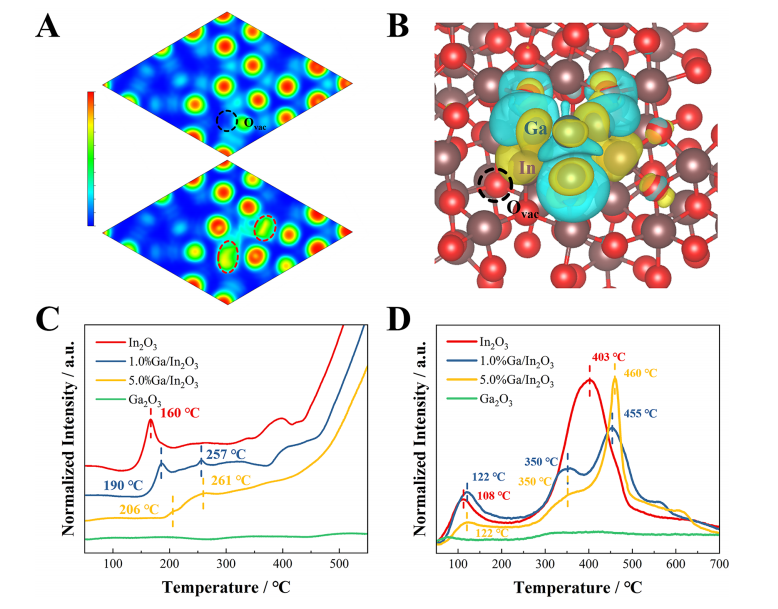
图4. 活性界面位点。(A)In2O3_D模型(上)和Ga4/In2O3_D模型(下)的电子局域函数(ELF)等高线图,(B)Ga4/In2O3_D模型中Ga4簇的电荷密度差异,黄色和蓝色等高面分别表示电子积累和电子耗尽,(C)原始In2O3、1.0%Ga/In2O3、5.0%Ga/In2O3和Ga2O3催化剂的H2程序升温还原(H2-TPR)和(D)O2程序升温脱附(O2-TPD)图谱。
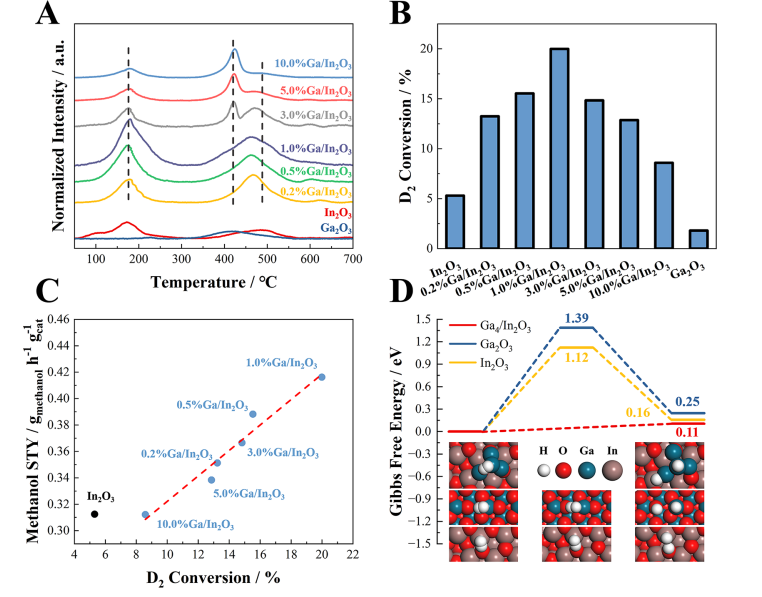
图5. H2解离吸附。(A)H2-TPD图谱,(B)200°C下原始In2O3、Ga2O3和Ga/In2O3催化剂的H2-D2同位素交换实验,(C)D2转化率与甲醇STY的关系,(D)Ga4/In2O3、Ga2O3和In2O3上H2解离吸附的结构和Gibbs自由能图谱。
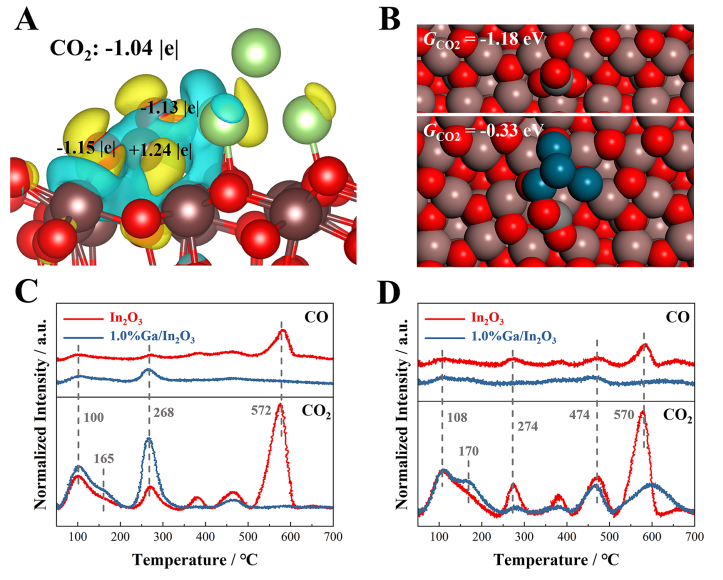
图6. CO2吸附与活化。(A)CO2在Ga4/In2O3_D模型的活性界面位点上的电荷密度差异,(B)CO2在In2O3_D模型上作为碳酸根(上)和在Ga4/In2O3_D模型的活性界面位点(下)的优化结构,(C)原始In2O3和1.0%Ga/In2O3催化剂的CO2-TPD和(D)CO-TPD图谱。
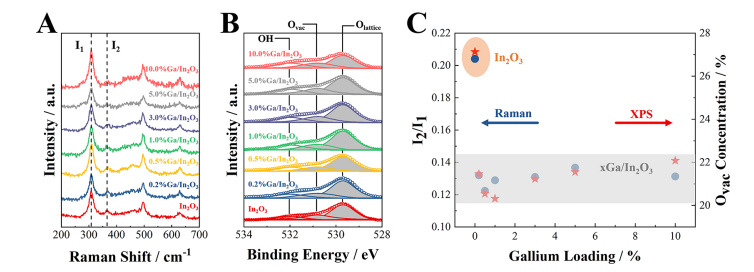
图7. 结构稳定性。(A)拉曼光谱,(B)原始In2O3和Ga/In2O3催化剂的O 1s XPS光谱,(C)根据拉曼光谱和O 1s XPS光谱的表面氧空位浓度与镓负载量的关系。
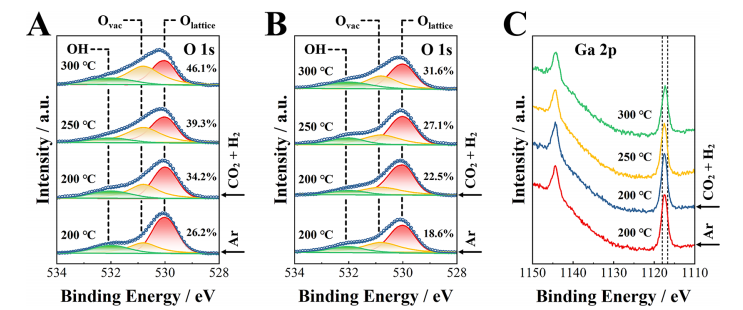
图8. 结构演变。准原位条件下的XPS光谱,O 1s光谱的准原位XPS图:(A)原始In2O3,(B)1.0%Ga/In2O3催化剂,(C)1.0%Ga/In2O3催化剂的Ga 2p光谱。
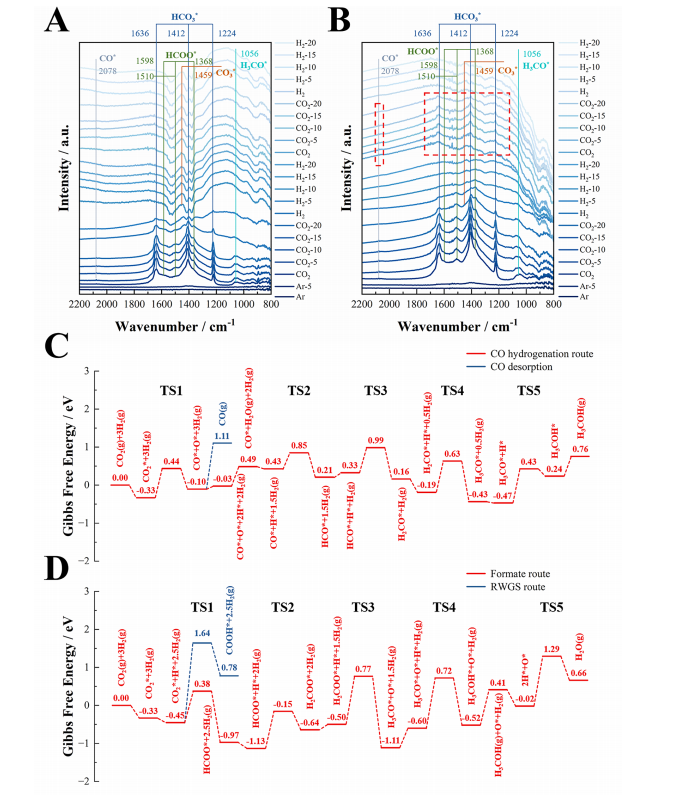
图9. 催化机理。原位漫反射红外傅里叶变换光谱(DRIFTS),200°C和0.1 MPa下的CO2加氢反应:(A)原始In2O3,(B)1.0%Ga/In2O3催化剂的原位DRIFTS光谱,(C)通过CO氢化、(D)甲酸盐途径和反向水煤气变换反应(RWGS)途径的Ga4/In2O3_D模型上CO2加氢制甲醇的Gibbs自由能图谱。
试验方法
X射线光电子能谱(XPS)
方法说明:XPS用于分析催化剂表面元素的化学态及电子状态,通过测量从样品表面发射的光电子的动能和数量,推断元素的价态和化学环境。
案例说明:本文通过XPS表征催化剂中的镓(Ga 2p)和氧(O 1s),确认了镓簇与氧化铟之间的电子相互作用,并研究了镓的氧化态变化。
X射线衍射(XRD)
方法说明:XRD用于分析催化剂的晶体结构和相组成,通过测量X射线在晶格中的衍射角度和强度,确定材料的晶体相和晶格结构。
案例说明:XRD分析揭示了不同镓负载量下In2O3催化剂的晶体结构变化,以及镓簇的形成和Ga2O3纳米晶体的生成。
程序升温还原(H2-TPR)
方法说明:H2-TPR用于研究催化剂中不同氧物种的还原行为,通过升温条件下的氢气还原反应,揭示催化剂的表面和体相氧的可还原性。
案例说明:本文通过H2-TPR实验确定了镓簇与氧化铟之间的界面氧还原特性,以及这些界面在促进CO2氢化反应中的作用。
程序升温脱附(CO2-TPD, CO-TPD)
方法说明:该技术用于分析气体在催化剂表面上的吸附和脱附行为,揭示催化剂对反应气体(如CO2和CO)的吸附能力及解吸温度。
案例说明:通过CO2-TPD和CO-TPD实验,本文研究了镓簇负载的In2O3催化剂对CO2和CO的吸附特性,进一步探讨了CO2氢化的机理。
拉曼光谱(Raman)
方法说明:拉曼光谱用于探测催化剂表面的振动模式,特别是氧化物中的表面氧空位和氧键振动。
案例说明:本文使用拉曼光谱量化了不同镓负载量的催化剂表面氧空位的浓度,分析了表面氧空位对催化剂稳定性的影响。
电子顺磁共振(EPR)
方法说明:EPR用于检测样品中未成对电子的存在,常用于表征氧空位等表面缺陷。
案例说明:在该研究中,EPR表征了镓掺杂催化剂中的表面氧空位,进一步揭示了镓促进CO2氢化的活性位点。
密度泛函理论计算(DFT)
方法说明:DFT是一种量子力学计算方法,用于模拟催化剂表面反应过程的能量变化和反应路径。
案例说明:本文通过DFT计算,模拟了镓簇与In2O3催化剂表面的相互作用,揭示了CO2和H2在催化剂界面上的吸附与解离过程。
▼▼▼
我们提供相关测试表征和理论计算服务,包括拉曼光谱、X射线衍射(XRD)和密度泛函理论(DFT)计算等服务。如果您有相关需求,可以联系我们。
添加下方微信好友,立即咨询选择我们的科研服务,带你发顶刊走在化学领域的最前沿电话/微信:13564914850创新思路
本论文的创新在于首次引入高度分散的镓簇作为氧化铟(In2O3)催化剂的助剂,用于二氧化碳加氢制甲醇反应。相比传统的铜基催化剂,镓簇与In2O3之间的强电子相互作用显著增强了催化剂的CO2转化率、甲醇选择性和长期稳定性。此外,本文通过多种表征技术和密度泛函理论(DFT)计算,揭示了镓簇与氧化铟表面氧空位的界面协同作用,明确了镓簇的促进机理,为非铜基催化剂的设计提供了新的思路和方向。
总结展望
本研究通过引入高度分散的镓簇成功提升了In2O3催化剂在CO2加氢制甲醇反应中的性能,显著提高了CO2转化率、甲醇选择性和长期稳定性。实验和理论计算表明,镓簇与氧化铟表面氧空位之间的界面协同作用是提高催化剂活性的重要因素。未来的研究可以进一步优化镓的负载量和分散度,探索其他非贵金属助剂的潜力。此外,还应关注反应条件下催化剂的结构演变及其与工业应用的契合度,以推动该类催化剂在实际碳捕集与利用(CCU)技术中的应用。
我是元素魔方科研服务
为多所高校、机构和企业提供专业科研支持
在节约科研成本的同时
还提供高效、全面的科研解决方案
我正好专业,你正好需要





















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











