







 广西大学团队对中国黑龙江省蜱类及其相关病毒进行研究。采用分子方法分类蜱虫样本,用下一代测序等评估蜱类病毒。鉴定出5种蜱类,获多个高质量reads,发现多种与已知病毒序列密切相关的新蜱传病毒,其对人畜健康意义待研究,研究为预防蜱传疫情提供依据。
广西大学团队对中国黑龙江省蜱类及其相关病毒进行研究。采用分子方法分类蜱虫样本,用下一代测序等评估蜱类病毒。鉴定出5种蜱类,获多个高质量reads,发现多种与已知病毒序列密切相关的新蜱传病毒,其对人畜健康意义待研究,研究为预防蜱传疫情提供依据。
研究简介
在世界范围内,蜱虫与各种人类和牲畜病原体的传播有关。大多数由蜱虫导致的感染发生在参与畜牧业的农民或在森林地区生活工作的人身上。这些传染病中最常见的有蜱传脑炎(TBE)、克里米亚-刚果出血热(CCHF)、发热伴血小板减少综合征(SFTS)和斑疹热。2009年,中国湖北和河南省的几个农民被蜱虫咬伤,他们后来死于一种不典型的出血热样疾病。该病原体是一种新型正支原体病毒,于2010年被确定为SFTS病毒(SFTSV)。2010年至2016年10月,中国有23个省份报告了SFTS病例。目前,蜱传病原体正日益威胁着全世界人类和动物的健康。因此,作为传染病预防策略的一部分,研究蜱虫媒介以及这些体外寄生虫的病毒谱很重要,可以更好地预测蜱虫传播疾病的潜在爆发。
2018年7月,广西大学动物科学技术学院Meng Fei研究团队在Ticks and Tick-borne Diseases发表了题为Virome analysis of tick-borne viruses in Heilongjiang Province, China的研究论文,旨在了解中国黑龙江省蜱类及其相关病毒的地理分布情况。文章描述了该研究团队采用分子方法对蜱虫样本进行分类,采用下一代测序和基于聚合酶链式反应的分析对大兴安岭四个代表性采样点的蜱类病毒进行评估。一共鉴定出5种蜱类,包括全沟硬蜱、草原革蜱、森林革蜱、长角血蜱和短血蜱。研究团队通过NGS测序从1102只蜱虫中获得了3,568,561个高质量reads。测序数据经质控后得到了302,540个reads,其中6577(2.16%)个reads注释到了病毒基因组上。系统发育分析表明,这些病毒序列与原病毒、静脉病毒、鹿蜱单胞病毒样病毒和荆门蜱病毒序列具有密切关系,但这些新鉴定的蜱传病毒对人畜健康的意义尚需进一步研究。该研究不仅为进一步研究蜱与蜱传病毒的关系,也为今后通过病媒控制预防蜱传疫情的爆发提供了依据。

研究结果:
在2015至2016年间,该研究团队从大兴安岭地区总共收集了1881只蜱虫,发现它们属于五个不同的种类:I. persulcatus(全沟硬蜱), H. longicornis(长角血蜱), H. concinna(短血蜱), D. nuttalli(草原革蜱) 和 D. silvarum(森林革蜱)。蜱虫的采集地点以及蜱虫在四个采样地点的分布情况分别如图1、表1所示。分析表明,I. persulcatus蜱虫的分布最普遍(48.9%),在所有四个采样地点均有分布。在双河和绰纳河两个自然保护区均发现了D. nuttalli(3.5%),H. longicornis(20.1%)和H. concinna(26.3%)仅在绰纳河自然保护区中发现。D. silvarum(1.2%)仅在双河自然保护区中发现。
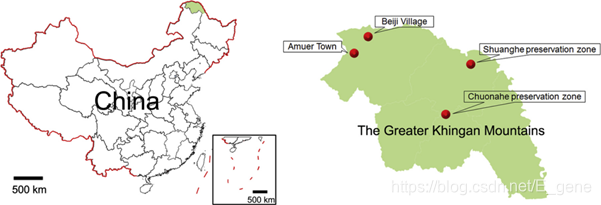
图1:蜱虫收集地点地图
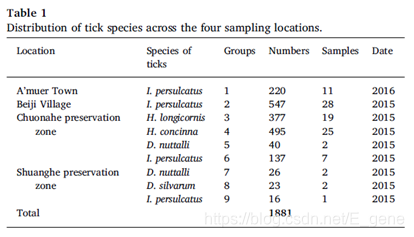
表1:四个采样地点的蜱类分布
通过Solexa测序获得了总计2030 Mb的原始reads。通过与从NCBI数据库获得的蜱虫基因组进行比较,且去除蜱虫序列后,总共剩下3,568,561个reads。 删除non-overlapping reads(7.85%)后总共得到3,288,385个overlapped reads,平均长度为134 bp。根据NCBI的细菌,真菌和病毒基因组数据库对这些clean数据进行初步分析,分别确定了395(0.011%),19,165(0.537%)和287,839(8.066%)个reads与已知的病毒,真菌和细菌序列相同。使用MetaGeneMark进行的基因预测显示,基于功能注释且在定义的临界值下,有304,540个reads显示了与已注释的微生物ORFs的同一性,而6,577个(2.16%,6,577 / 304,540)reads显示了与病毒性ORFs的同一性。基于最显著的BLASTn相似度(E值≤10e-5),病毒序列分为18个病毒家族(图2)。大约98.42%(6,473/6,577)的reads被注释为跨越12个病毒科的脊椎动物病毒,包括异病毒科、奈罗病毒科、横纹虫病毒科、逆转录病毒科、环布尼亚病毒科、异戊病毒科、疱疹病毒科、黄病毒科、诺达病毒科、小核糖核酸病毒科、乳头瘤病毒科和一个未分类的病毒科,包括荆门蜱病毒(JMTV)、鹿蜱单胞病毒样病毒(DTMV)、肩周骨细线虫相关病毒、潘多拉病毒、武汉虱蝇病毒、亚人类病毒、禽流感病毒和蜱传四足病毒样病毒。
6,577个病毒组reads被组装成91个contigs。正布尼亚病毒样序列占所有contigs的60%以上,其中30条contigs注释到SBV, 28条注释到BTPV, 14条注释到DTMV, 19条注释到JMTV。 Solexa测序结果也通过使用这些脊椎动物病毒特异性引物的巢式PCR进行了验证(表2)。结果表明,从不同地点采集的属于同一种的蜱,其PCR阳性病毒谱不同,但来自同一地点的不同种蜱也表现出不同的病毒组谱。
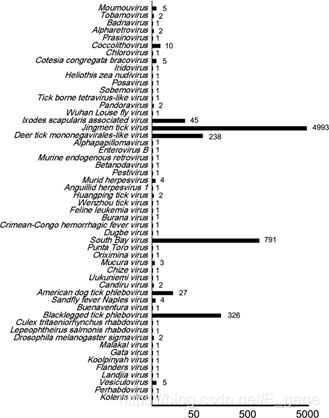
图2:从样品获得的病毒reads数目概览
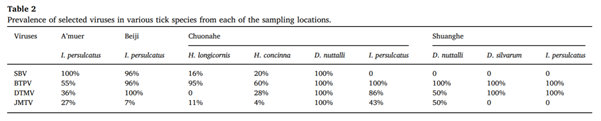
表2:从每个采样地点的各种蜱类中选定的病毒的流行情况
奈罗病毒科属于布尼亚韦拉斯目,由12个物种组成,其中许多是蜱传病毒。2014年对北美蜱进行的病毒组研究确认了SBV,并将其归类为一种属于奈罗病毒家族的新病毒。与之前鉴定的蜱媒原状病毒种类相比,SBV具有高度的差异性。在当前的研究中,分别从几个SBV阳性样品中扩增了L和S片段的12708-bp和1092-bp片段,并进行了测序。用来自所有12种原发性病毒代表性序列对所得序列进行系统发育分析,结果表明,从Beiji采集的样本中提取的一个序列(因此被命名为Beiji奈罗病毒)与所述的任何一种原发性病毒没有聚集在一起(图3A和B)。
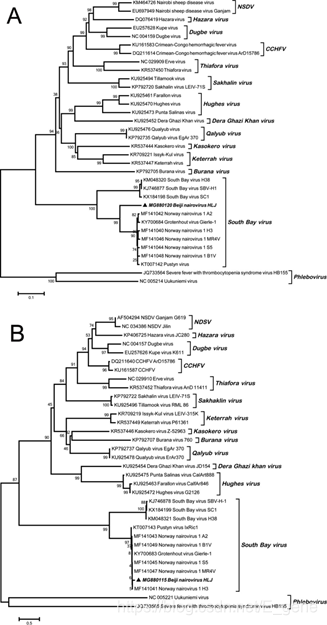
图3:(A)系统发育分析,基于来自Beiji奈罗病毒和其他代表性蜱虫传播病毒的RNA依赖性RNA聚合酶基因(L段)的12,708-bp区域。(B)系统发育分析,基于Beiji奈罗病毒与其他代表性蜱虫传播病毒的核衣壳蛋白基因(S区段)的1092-bp区域。使用最大似然模型的邻域连接(NJ)方法生成树。分析包括1000个引导重复。分支上方的数字表示NJ引导值。粗体三角形表示当前研究中获得的Beiji奈罗病毒序列。
静脉病毒属也属于布尼亚病毒属,该属的病毒显示出与正腮腺病毒属病毒的基因组同一性。基于NGS数据,设计特定的引物,通过基于RT-PCR的扩增检查样品中BTPV的发生率。以L片段540-bp片段为靶点,对所有蜱虫标本进行巢式RT-PCR,证实了Solexa测序结果。如表2所示,在所测试的蜱类样本中,BTPV的流行率很高,I. persulcatus(全沟硬蜱)的患病率为55%,D. nuttalli(草原革蜱)和D. silvarum(森林革蜱)的患病率为100%。对从PCR阳性样品中扩增出静脉病毒序列L片段的2003 bp片段并进行测序。序列比较表明,所得序列与挪威静脉病毒1、BTPV-1、BTPV-2和BTPV-3的核苷酸序列一致性分别为81%、78%、76%和70%。基于L片段序列的系统发育分析表明,该研究鉴定的Beiji奈罗病毒序列属于蜱传静脉病毒家族,但与尤库尼米病毒群和SFTS群分离,形成了一个单系支链(图4)。在I. persulcatus(全沟硬蜱)样本中DTMV的患病率为36-100%,在H. longicornis(长角血蜱)中为0%,在H. concinna(短血蜱)中为28%,在D. nuttalli(草原革蜱) 中为50-100%,在D. silvarum(森林革蜱)中为100%(表2)。
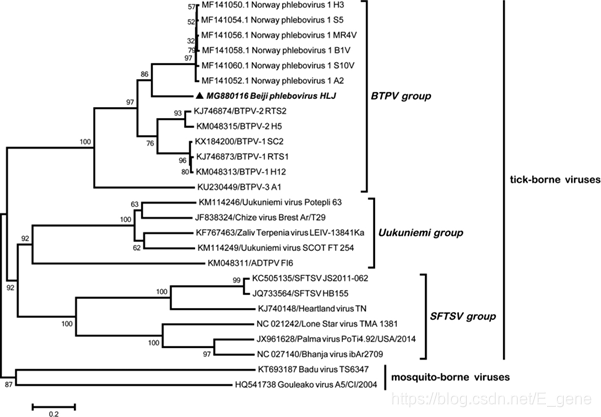
图4:基于RNA依赖的RNA聚合酶基因2003 bp区域的Beiji奈罗病毒和其他代表性蜱虫传播病毒的系统发育分析。 该树是使用邻域连接(NJ)方法和最大复合似然模型生成的。分支上方的数字表示NJ引导值。粗体三角形表示当前研究中获得的Beiji病毒序列。
通过PCR从NGS阳性样品中获得了来自DTMV的L区段(命名为DTMV HLJ)的3985bp的片段。 系统发育分析表明,DTMV HLJ序列与DTMV株DTM1和F13具有72%的同源性。在单反病毒株内形成了一个独立的单系分支(图5)。
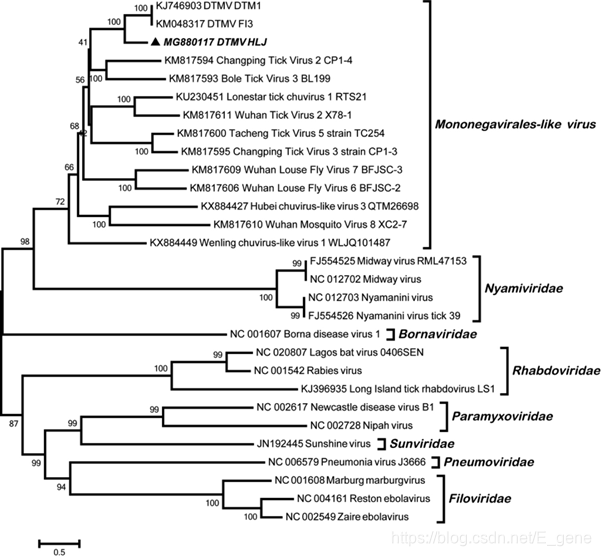
图5:基于RNA依赖的RNA聚合酶基因的3985 bp片段,对鹿蜱单胞病毒样病毒(DTMV)HLJ株和其他代表性病毒进行系统发育分析。该树是使用邻域连接(NJ)方法和最大复合似然模型生成的。分支上方的数字表示NJ引导值。粗体三角形表示当前研究中鉴定出的DTMV菌株HLJ。
JMTV有一个分段的RNA基因组,来自一个未分割的黄病毒科病毒,广泛分布于中国各地的蜱类种群中。使用针对NS5(片段1)的特异性引物对所有样本进行基于RT-PCR的筛选,结果表明,在I. persulcatus(全沟硬蜱)样品中的JMTV患病率为0–43%,在H. longicornis(长角血蜱)中为10%,在H. concinna(短血蜱)中为4%,在D. nuttalli(草原革蜱) 中高达100%(表2)。然而,在D. silvarum(森林革蜱)样品中未检测到JMTV(表2)。通过PCR从一些PCR阳性样品中获得NS5(片段1)的1298bp片段和NS3(片段3)的1865bp片段,随后进行测序。系统发育分析表明,在两个系统发育树中,JMTV序列均与黄病毒和类黄病毒序列聚集在一起(图6A和B)。
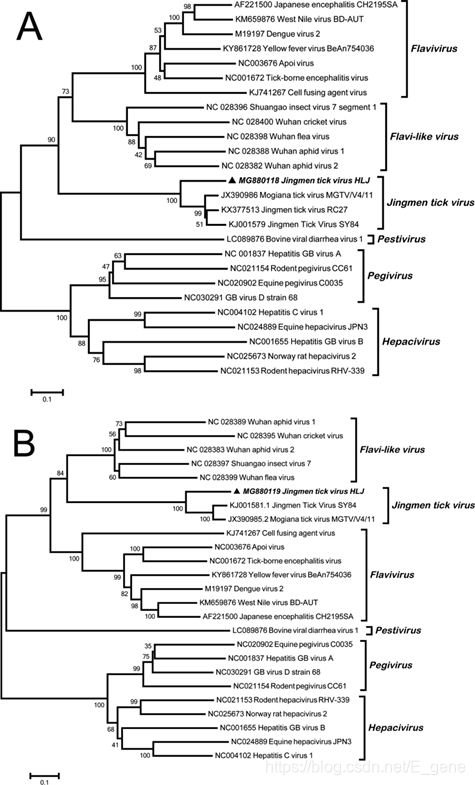
图6:荆门蜱病毒(JMTV)新株HLJ与黄病毒属(黄病毒科)代表性病毒的系统发育分析。基于NS5(片段1)基因(A)的1298bp片段和NS3(片段3)基因(B)的1865bp片段构建了系统发育树。使用最大似然模型的邻域连接(NJ)方法生成树。分支上方的数字表示NJ引导值。粗体三角形表示当前研究中确定的JMTV株HLJ。
结论:
作者利用在大兴安岭获取的蜱虫样本,鉴定出五种蜱类,并对其进行病毒组分析。结果表明,有几种病毒可能是新的蜱传播病毒。此研究未检出东北地区自然疫源地病毒,包括TBEV、STSFV、CCHFV和NSD病毒,作者认为这可能是地理隔离、蜱虫样本量和采样时间的原因。目前尚不清楚这些新发现的病毒是否能通过血液喂养传播给脊椎动物宿主,因此,它们对人类和动物健康的意义还需要进一步研究。此外,需要利用更大地理区域的样本进行研究,以便更好地了解蜱类分布和蜱传病毒的流行情况,这将有助于预防和控制蜱传疾病。
参考文献:
Meng F , Ding M , Tan Z , et al. Virome Analysis of Tick-borne Viruses in Heilongjiang Province, China[J]. Ticks and Tick-borne Diseases, 2018.
文中病毒组研究方法:
- 该研究结合蜱虫分类与下一代测序从各类蜱虫携带病毒中挖掘病毒组学数据。
- 在蜱虫样本中提取RNA,反转录成cDNA并扩增,用于后续测序。
- 使用BLASTx和BLASTn分析,将所得序列数据与非冗余和病毒参考GenBank数据库进行比对。
- 利用MEGA 6生成系统发育树,并利用Clustal W进行核苷酸序列比对。
技术文章解读:点击查看原文


















 6674
6674

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









