







研究方向非生信领域,只是学习一下模型,所以有关生物(结论)的部分没有翻译
文章目录
摘要
确定介导前列腺癌临床侵袭性表型的分子特征仍然是一个主要的生物学和临床挑战1,2。应用于生物医学问题的机器学习模型的可解释性方面的最新进展可能有助于发现和预测临床癌症基因组3–5。在这里,我们开发了P-NET——一种生物信息深度学习模型 ——根据治疗耐药状态对前列腺癌患者进行分层,并通过完整的模型解释性评估治疗靶向性治疗耐药的分子驱动因素。我们证明P-NET可以使用分子数据预测癌症状态,其性能优于其他建模方法。此外,P-NET的生物学解释性揭示了已建立和新的分子改变候选基因,如MDM4和FGFR1,它们与预测晚期疾病有关,并在体外得到验证。从广义上讲,生物信息完全可解释的神经网络能够在前列腺癌中进行临床前发现和临床预测,并且可能具有跨癌症类型的普遍适用性。
引言
随着分子分析技术的发展,在过去的十年里,观察数百万癌症患者及其肿瘤的基因组、转录和其他特征的能力显著增长。具体来说,在前列腺癌中,与临床注释相关的丰富的分子图谱数据的可用性,使许多个体基因、通路和复合物得以发现,它们促进了致命的去势抵抗性前列腺癌(CRPC),这导致了对这些个体特征进行生物学研究和临床评估,以预测效用1,2,6 - 12。然而,这些分子特征与它们对疾病进展、耐药性和致死结果的预测和生物学贡献之间的关系仍在很大程度上没有特征。
在开发预测模型时,有各种各样的潜在方法,尽管每种方法都需要在准确性和可解释性上进行权衡。在转译癌症基因组学中,预测模型的可解释性是至关重要的,因为有助于模型预测能力的特性不仅可以告知患者护理,还可以提供对潜在生物学过程的洞察,以促进功能研究和治疗靶向。logistic回归等线性模型往往具有较高的可解释性,但预测性能较差,而深度学习模型往往具有较低的可解释性,但预测性能较高13,14。使用典型的全连接密集深度学习方法来构建预测模型也可能导致过拟合,除非网络是很好的正则化的,而且这种模型有计算成本高且难以解释的趋势15。
在给出完整模型的情况下,我们努力寻找更精简的架构和稀疏网络,结果表明稀疏模型可以降低存储需求,提高计算性能16 - 18。然而,寻找这样的稀疏模型可能是一个挑战,因为典型的训练-剪枝-再训练周期通常是计算昂贵的,最近的研究表明从头构建稀疏模型可能更容易19。此外,为了提高深度学习模型的可解释性以及解释模型决策的需要,开发了多种归因方法,包括LIME20、DeepLIFT13、DeepExplain21和SHAP22,这可以用来增强深度学习的可解释性,并理解模型是如何处理信息和做出决策的。
同时,稀疏模型开发和归因方法的进步为深度学习模型的开发提供了信息,该模型使用受生物系统启发的定制神经网络架构来解决生物问题。例如,可视神经网络被开发用来模拟基因相互作用对酵母细胞生长的影响(DCell),以及癌症细胞系与疗法的相互作用(DrugCell)3,5。在多形性胶质母细胞瘤中,一种与通路相关的稀疏深度神经网络(PASNet)使用一种扁平的通路来预测患者预后23。然而,生物学信息神经网络是否能够加速具有翻译潜力的生物发现,并同时使临床预测模型成为可能,这在很大程度上是未知的。在这里,我们假设一个基于稀疏深度学习架构的生物信息深度学习模型,与已建立的模型相比,生物信息编码和可解释算法的结合将获得更好的预测性能,并揭示前列腺癌治疗耐药性的新模式,具有翻译意义。
结果
我们开发了一种深度学习预测模型,将之前生物学上建立的层次知识整合到神经网络语言中,以根据前列腺癌患者的基因组谱预测其癌症状态。利用3007条经过精心策划的生物通路构建了一个路径感知的多层层次网络(P-NET)(方法)。在P-NET中,个体的分子剖面被输入到模型中,并通过加权链接分布在代表一组基因的节点层上(图1,扩展数据图1)。随后的网络层编码了一组抽象水平不断提高的路径,较低的层次代表精细的途径,较晚的层次代表更复杂的生物途径和生物过程。不同层之间的连接被限制在已知的编码特征、基因和路径之间的亲子关系,因此,网络的设计是面向可解释性的。

图1 |可解释的生物信息深度学习。 P-NET是一种神经网络体系结构,它将不同的生物实体编码成一种神经网络语言,并在连续的层次(即患者档案、基因、路径、生物过程和结果的特征)之间定制连接。在这项研究中,我们主要关注处理突变和拷贝数的改变。经过训练的P-NET提供了每一层节点的相对排序,以便提供生物假设的生成。实线显示了从输入到生成结果的信息流,虚线显示了计算不同节点重要性得分的方向。候选基因被验证以了解其功能和作用机制。
- 我们对1,013例前列腺癌(333例CRPCs和680例原发癌)进行了P-NET训练和测试(补充表1-5),
分为80%训练,10%验证和10%测试,利用体细胞突变和拷贝数数据预测疾病状态(原发或转移性疾病)(方法)。经过训练的P-NET优于典型的机器学习模型,包括线性和径向基函数支持向量机、逻辑回归和决策树 ( ROC曲线下面积(AUC) = 0.93,精度-召回曲线下面积(AUPRC) = 0.88,准确度= 0.83)(图2、扩展数据图2,补充表6、7,方法)。在P-NET中加入额外的分子特性是可行的(例如,融合)但不影响模型的性能在这个特定的预测任务(扩展数据图3、4)。
- 此外,我们评估了稀疏模型是否具有不同于密集全连接深度学习模型的特征。
我们在训练集上使用与P-NET模型相同的参数数训练密集模型,样本数从100到811(占总样本数的80%)按对数递增。P-NET模型的平均性能(由AUC决定)在所有样本量上都高于密集模型,这种差异在较小的样本量(高达500)上具有统计学意义。(例如,与在155个样本上训练的密集网络相比,P- net的五倍交叉验证的平均AUC显著更高,P = 0.004)(图2c,扩展数据图5a-e;统计检验结果如补充表8所示)。此外,与P-NET的神经元数量和层数相同,但参数数量(1400万)更多的稠密网络的性能也较差(扩展数据图5f)。
- 接下来,我们对模型的预测方面进行了外部验证,使用了两个额外的前列腺癌验证队列,一个原发性24和一个转移性25(样本标识符列于补充表4,5;方法)。
经过训练的P-NET模型正确分类了73%的原发肿瘤和80%的转移性肿瘤,表明该模型可以推广到未见过的样本,具有足够的预测性能(图2b)。我们假设原发肿瘤样本被P-NET错误地归类为去势抵抗转移性肿瘤的患者实际上可能有更坏的临床结果。与P- net评分低的患者相比,P- net评分高的患者被误诊为耐药性疾病的患者明显更容易出现生化复发(P = 8 × 10−5;log-rank检验),表明对于原发性前列腺癌患者,P-NET评分可用于预测潜在的生化复发(图2d,补充表9)。

图2 | P-NET的预测性能。
a、 当在测试集上进行测试时,P-NET在AUPRC(括号中显示的值)方面优于其他模型(n=204,来自亚美尼亚等数据集8)。径向基函数。
b、 当使用两个独立的外部验证组合24,25进行评估时,P-NET实现了73%的真阴性率(TN)和80%的真阳性率(TP),表明它可以推广到对未知样本进行分类。FN,假阴性率;FP,假阳性率。
c、 与具有相同数量参数的密集全连接网络相比,P-NET在样本数量较少的情况下实现了更好的性能(以五次交叉验证拆分的平均AUC衡量)。实线表示平均AUC,条带表示平均值±标准偏差(n=5次实验)。对于500以下的所有样本量,性能差异在统计学上具有显著性(*P<0.05,单侧t检验)(方法)。
d、 原发性前列腺癌和高P-NET评分HPS(被P-NET错误分类为耐药样本)患者与低P-NET评分LPS患者相比,表现出更大的生化复发(BCR)倾向,后者倾向于无进展生存(P=8×10)−5.对数秩检验,双侧)。这表明P-NET模型可能有助于临床患者分层和预测潜在BCR(原始数据包含在补充表9中)。LPS,低P-NET评分;HPS,高P-NET分数。
- 为了理解不同特征、基因、途径和生物过程之间的相互作用,以及研究从输入到结果的影响路径。
经过训练后,我们用完全可解释的层将P-NET的整个结构可视化(图3)。在总体分子改变中,拷贝数变化比突变更能提供信息,这与之前的报道一致26。

图3 |检查和解释P-NET。P-NET内层的可视化显示了每层中不同节点的估计相对重要性。 最左侧的节点表示特征类型;第二层的节点代表基因;下一层代表更高层次的生物实体;最后一层表示模型结果。颜色较深的节点更重要,而透明节点表示每个层中未展开节点的剩余重要性。使用Sankey图描述了特定数据类型对每个基因重要性的贡献。例如,AR基因的重要性主要由基因扩增驱动,TP53的重要性由突变驱动,PTEN的重要性由缺失驱动。
- 此外,P-NET选择了一个与分类相关的通路层次(在P-NET训练的3007个通路中),包括细胞周期检查站、翻译后修饰(包括泛素化和SUMOylation)和RUNX2和TP53的转录调节。
细胞周期途径的多个成员在功能上与转移性前列腺癌有关,特别是在耐药情况下的功能审问。泛素化和sumo化途径有助于调节多种肿瘤抑制因子和癌基因,包括AR29,这些途径的失调已被证实与临床前模型中前列腺癌的起始和进展有关30。RUNX2是一种调节细胞增殖的成骨转录因子,与前列腺癌患者的转移性疾病相关31。
- 为了评估有助于模型预测的特定基因的相对重要性,我们检查了基因层,并使用DeepLIFT归因法获得基因的总重要性得分(Methods)13。
高排名的基因包括AR、PTEN、RB1和TP53,它们是已知的前列腺癌驱动因子,以前与转移性疾病相关。此外,较少预期基因的改变,如MDM4、FGFR1、NOTCH133和PDGFA,对预测性能有很大的贡献(扩展数据图6、7)。
- 为了了解经过训练的P-NET的行为,我们检查了网络中每个节点的激活情况。
这里的激活表示给定输入的特定节点的签名结果,并测试该激活是否随着输入样本类别的改变而改变(主要的还是转移的)(方法)。我们观察到,节点激活的差异在越高的层次上越高,并且在每一层中越集中在高等级的节点上(扩展数据图8)。例如,与抗性样品相比,当P-NET被给予初级样本时,层H3的节点的激活分布不同(扩展数据图8c) 。因此,可以利用P-NET的可解释架构来理解输入信息是如何通过层和节点进行转换的,从而进一步理解所涉及的生物实体的状态和重要性。
- 通过对P-NET训练模型中多个层次的评估,我们观察到TP53相关生物学中有助于CRPC的收敛。
追踪TP53相关通路与基因水平的相关性,TP53和MDM2在前列腺癌疾病进展中的作用已被确定32,34–40,我们还观察到MDM4的改变,这些改变对这种网络融合起到了实质性的作用。MDM4可通过结合并掩盖转录激活域40抑制野生型TP53表达,尽管其在前列腺癌治疗耐药性中的作用尚不完全确定41。
- 我们进一步研究了MDM4在临床样本和功能模型中的分布。
与原始样本相比,耐药样本中MDM4的高扩增更为普遍(χ2-Yates系数=40.8251,P<0.00001)。AR、TP53和MDM4基因的改变如图4a所示。在LNCaP细胞中使用17255开放阅读框(ORF)进行的全基因组功能增益临床前筛查中,MDM4过度表达与对恩扎鲁胺(一种用于CRPC42患者的第二代抗雄激素药物)的耐药性显著相关(图4b)。然后,我们使用CRISPR–Cas9在多个前列腺癌细胞系中靶向MDM4(方法)。
与阴性对照组相比,使用两种不同的单导向RNA(SGRNA)去除MDM4后,前列腺癌细胞的增殖显著减少(P<0.0001;t检验)(图4c;补充数据1)(扩展数据图9,补充数据2)。这表明MDM4的选择性靶向治疗在TP53野生型晚期前列腺癌患者中可能是可行的。因此,我们试图研究突变型和野生型TP53对前列腺细胞系中MDM4的抑制作用。与TP53突变细胞系43相比,具有野生型TP53的前列腺细胞对MDM4选择性抑制剂RO-5963(其也抑制MDM2)更敏感(图4d;方法)。总的来说,经过训练的P-NET模型的多个层面上的p53通路失调的聚合确定了涉及MDM4的特定脆弱性,MDM4可以作为治疗靶点。
讨论
从广义上讲,P-NET利用了一种生物信息而非任意过度参数化的预测架构。因此,P-NET显著减少了学习参数的数量,从而提高了可解释性。与其他机器学习模型(包括密集网络)相比,P-NET中的稀疏结构具有更好的预测性能,并且可能适用于其他类似任务。
P-NET在前列腺癌患者分子队列中的应用证明
- 模型性能可预测原发性前列腺癌患者群体中的临床侵袭性疾病
- 聚合的生物学过程有助于转移性前列腺癌临床表型,在分子分层人群中具有新的治疗策略
此外,P-NET提供了一种简单的方法来整合多个不同权重的分子特征(例如突变、拷贝数变化和融合等),以反映其在预测最终结果中的重要性,以前需要对每个特征采用不同的统计方法才能发现癌症基因44,45。
更重要的是,P-NET提供了一个框架,用于使用神经网络语言编码分层先验知识,并将这些分层转化为计算模型,既可用于预测,也可用于临床遗传学背景下的生物发现。具体而言,P-NET基于患者的基因组图谱准确预测晚期前列腺疾病,并具有预测潜在生化复发的能力。P-NET体系结构的可视化使我们能够对涉及的生物途径和过程进行多层次的观察,这可能会指导研究人员开发关于癌症进展中潜在生物过程的假设,并将这些发现转化为治疗机会。具体而言,P-NET重新发现了与CRPC相关的已知基因,如AR、PTEN、TP53和RB1。此外,P-NET将MDM4确定为该临床背景下的一个相关基因,这已通过实验验证,并可能为转移性前列腺癌基因组分层(TP53野生型)患者使用MDM4选择性抑制剂提供信息。
尽管P-NET为结果预测和假设生成提供了框架,但该模型在使用前仍需要调整和训练。与所有深度学习模型一样,最终训练的模型在很大程度上取决于用于训练模型的超参数。此外,P-NET以硬编码方式对网络内的生物路径进行编码,这使得模型依赖于用于构建模型的注释的质量。使用利用其他硬编码生物先验(如KEGG和基因本体)或用户指定的特定生物模块的模型可以进一步指导模型开发和功能评估。最后,计算方面的进步可能使这种方法能够在患者特定的精确肿瘤学示意图中使用,并与患者特定的模型系统配对,以进行直接可比的实验评估。因此,这种方法在不同组织学和临床背景下的可移植性需要进一步评估。
总之,P-NET是一种生物学信息丰富的深层神经网络,能够准确地将去势抵抗的转移性前列腺癌与原发性前列腺癌进行分类。可视化训练模型产生了前列腺癌转移机制的新假设,并为分子分层前列腺癌患者群体的临床转化提供了直接潜力。生物引导神经网络是一种通过建立机械预测模型将癌症生物学与机器学习相结合的新方法,为生物发现提供了一个平台,可广泛应用于癌症预测和发现任务。
方法
1 - P-NET设计
我们介绍了P-NET,这是一种人工神经网络,具有生物信息、简约的结构,能够根据前列腺癌患者的基因组图谱准确预测其转移。
P-NET是一种在节点和边上有约束的前馈神经网络。在P-NET中,每个节点编码一些生物实体(例如,基因和通路),每个边表示相应实体之间的已知关系。节点上的约束允许更好地了解不同生物成分的状态。边上的约束允许我们在不增加边数的情况下使用大量节点,这导致与具有相同数量节点的全连接网络相比,参数数量更少,因此计算量可能更小。
该结构是使用Reactome通路数据集构建的。整个Reactome数据集被下载和处理,形成一个由五层通路、一层基因和一层特征组成的分层网络。该稀疏模型的权重略高于71000,每层节点数分布如扩展数据图1e所示。具有相同节点数的密集网络将具有超过2.7亿个权重,其中第一层包含超过94%的权重。一个包含稀疏层和密集层的混合模型仍然包含超过1400万个权重。密集权重的数量计算为 w w w l l l= n n n l l l×( n n n l − 1 l-1 l−1+1),其中 w w w l l l是每层 l l l的权重。 n n n l l l是同一层的节点数。请注意,P-NET模型不绑定到特定架构,因为模型架构是通过读取用户通过基因矩阵转置文件格式(.gmt)文件提供的模型规范自动构建的,并且用户可以提供自定义路径、基因集和具有自定义层次结构的模块。
P-NET的节点、层和连接的含义通过精心设计的体系结构和对网络连接的一系列限制进行编码。输入层表示可以测量并输入网络的特征。第二层代表一组感兴趣的基因。更高的层次代表手动管理的路径和生物过程的层次结构。P-NET的第一层通过一组一对一的连接连接到下一层,下一层中的每个节点恰好连接到输入层的三个节点,表示突变、拷贝数放大和拷贝数删除。与全连接网络相比,该方案在第一层中的权值数量要少得多,并且连接矩阵的特殊模式使训练更有效。第二层被限制为具有反映基因通路关系的连接,如Reactome通路数据集所策划的。连接由掩码矩阵M编码,掩码矩阵M乘以权重矩阵W,以将Reactome路径数据集中不存在的所有连接归零。对于下一层,设计了一个类似的方案来控制连续层之间的连接,以反映Reactome数据集中存在的真实父子关系。每一层的输出计算为 y y y= f f f[(M*W)Tx+b],其中 f f f是激活函数,M是掩模矩阵,W是权重矩阵,x是输入矩阵,b是偏置向量,*是哈达玛积(参见扩展数据图1a-c)。每个节点的激活保持在[−1,1]通过应用tanh函数 f f f= t a n h tanh tanh=(e2x− 1) / (e2x+1)到节点的加权输入。结果层的激活通过sigmoid函数σ=1/(1+e-x)计算。
为了让每一层本身都有用,我们在每个隐藏层之后添加了一个带有sigmoid激活的预测层。与图1e所示的第一层扩展数据相比,P-NET在后面的层中每层的节点数量较少。由于在后一层中使用较少的权重来拟合数据更具挑战性,因此在优化过程中,我们在后一层结果中使用了较高的损失权重。网络的最终预测是通过取所有层结果的平均值来计算的,扩展数据如图1d所示。学习率初始化为0.001,并在每50个阶段后积极降低,以实现平滑收敛。因为我们有一个不平衡的数据集,所以我们根据训练集中的偏差对类进行不同的加权,以减少对一个类的网络偏差。使用Adam优化器对模型进行训练,以减少二元交叉熵损失函数 H H H= - 1 N \frac{1}{N} N1 ∑ \sum ∑ y y y i i i. l o g log log( p p p( y y y i i i) ). l o g log log( 1 − p 1-p 1−p( y y y i i i) ),其中 y y y i i i是样本 i i i的标签, p p p( y y y i i i) 是样本 i i i具有转移癌的概率,使用sigmoid函数σ计算, N N N是样本总数。经验上,我们发现除了Adam之外,使用自适应学习率可以使收敛更加平滑,并提高预测性能。我们检查了不同的基于梯度的属性方法来对所有层中的特征进行排序,并选择使用DeepExplain库中实现的DeepLIFT方案。
DeepLIFT是一种基于反向传播的归因方法,用于为每个特征分配样本级别的重要性分数。在这项工作中,我们感兴趣的是为每个层中的每个节点分配分数。给定某个样本、特定目标
t
t
t和一组层节点
x
1
s
x^s_ 1
x1s,
x
2
s
x^s_ 2
x2s,
x
3
s
x^s_ 3
x3s, …,
x
n
l
s
x^s_ {n_l}
xnls 其中
n
l
n_l
nl是特定层l中的节点数,DeepLIFT根据目标激活
t
–
t
0
t–t_0
t–t0中的差异计算每个节点的重要性得分
C
i
l
,
s
C^{l,s}_ i
Cil,s,使得该差异等于所有节点计算得分的总和。也就是说,目标激活的差异由以下公式得出:
Δ
t
=
t
−
t
0
\Delta t=t-t_0
Δt=t−t0
等于由某个样本S馈送时所有节点得分的总和。也就是说,
Δ
t
=
∑
C
i
l
,
s
\Delta t=\sum C^{l,s}_i
Δt=∑Cil,s
我们使用DeepExplain实现的DeepLIFT的“重缩放规则”来计算所有层中所有节点的样本级别重要性。更多详情见参考文献13。为了计算总的节点级重要性
C
i
l
C_i^l
Cil,我们对所有
n
s
n_s
ns测试集样本的样本级重要性得分进行了汇总。
C
i
l
,
s
=
∣
∑
i
=
1
n
s
C
i
l
,
s
∣
C^{l,s}_i=|\sum_{i=1}^{n_s} C^{l,s}_i|
Cil,s=∣i=1∑nsCil,s∣
请注意,这是一个绝对分数(始终为正值),用于衡量某个节点对结果的影响。然而,相应节点i的激活可以是正的或负的。
为了减少某些节点(属于过多路径成员的节点)的过度注释所引入的偏差,我们使用考虑每个节点连通性的图通知函数 f f f调整了DeepLIFT分数。如果节点度大于节点度的平均值加上5 σ σ σ,则重要性得分 C i l C_i^l Cil除以节点度 d i l d_i^l dil,其中 σ σ σ是节点度的标准偏差。
d i l = f a n ‾ i n i l + f a n ‾ o u t i l d^l_i=fan\underline\space in^l_i + fan\underline\space out^l_i dil=fan inil+fan outil
a d j u s t e d ‾ C i l = f ( x ) = { C i l d i l , d i l > μ + 5 σ C i i , o t h e r w i s e adjusted\underline\space C_i^l=f(x)=\left\{\begin{matrix}\frac{C^l_i}{d^l_i},d^l_i>μ+5σ\\C_i^i,otherwise \end{matrix}\right. adjusted Cil=f(x)={dilCil,dil>μ+5σCii,otherwise
2 - P-NET训练和评估
为了验证所开发模型的实用性,我们训练P-NET根据前列腺癌患者的基因组图谱预测其癌症状态(原发性/转移性)。
我们对1013名患者进行了肿瘤或ermline匹配的全外显子组测序,以及相应的体细胞突变和拷贝数改变,这些都是通过统一的计算管道准备的,用于协调体细胞突变的产生(在本研究中注释为“亚美尼亚等人”队列)。这些突变在基因水平上被集中在非同义突变上,以与先前在前列腺癌全外显子数据集上的突变意义一致,排除沉默、内含子、3 '非翻译区(UTR)、5 ’ UTR、RNA和长基因间非编码RNA (lincRNA)突变。每个基因的拷贝数改变是根据所谓的片段级拷贝数分配的,强调高增益和深度缺失,排除单拷贝扩增和缺失,如GISTIC2.0定义的,由源数据类型生成。
对于涉及RNA数据(融合、表达)的二次分析,来自亚美尼亚等队列亚群的大量完整转录组(在这些数据可用的情况下)来自其来源研究(TCGA的n=455,SU2C-PCF consortia的n=204),用于RNA序列的统一比对和定量。从TCGA(ISB-CGC;https://isb-cgc.appspot.com/)和来自SU2C的CRAM(来自AmazonS3 bucket,dbGaP登录代码,phs00915.v2.p2),然后使用samtools fastq转换为FASTQs。在SU2C样本同时具有转录组捕获和polyA测序的情况下,转录组捕获用于优化融合检测,作为这些数据的主要用途。适配器用cutadapt v2.2修剪,读数使用STAR-aligned v2.7.2b。STAR-aligned的bam文件被传递到RSEM中,以使用GENCODE release 30基因注释生成基因水平的转录本计数和百万分之一转录本(TPM)定量,并转移到GRCh37。STAR嵌合连接被提供给STAR-Fusion v1.7.0在kickstart模式下调用fusions50。融合调用被过滤到那些在癌症基因普查中被归类为癌基因或融合基因的调用。为了测试基于RNA的融合输入的模型灵活性,作为二次分析,我们还开发了经过训练的P-NET模型,用于预测癌症状态,包括融合或拷贝数状态的不同定义(扩展数据图3、4)。
使用平均AUC、AUPRC和F1分数测量预测性能。报告了测试拆分和交叉验证设置的相应措施。输入数据分为测试集(10%)和开发集(90%)。开发集进一步划分为与测试集大小相同的验证集,其余样本保留用于训练。对于交叉验证实验,开发数据集按标签类别划分为五层,以说明数据集中的偏差。外部验证结果由在主数据集上训练并在两个独立的外部验证数据集上测试的模型生成。为了缓解主数据集中的偏差问题,我们在从主数据集中抽取的两个平衡子样本上训练了两个模型。对两个模型的预测分数进行平均,以在两个外部验证数据集上生成最终预测。GitHub上提供了建议系统的实施以及可再现的结果(https://github.com/marakeby/pnet_prostate_paper)。
3 - 统计分析
P-NET和其他模型之间ROC曲线下面积的变化使用DeLong test52进行测试。使用FDR对多假设检验的P值进行校正。对于其他分数,包括AUPRC、准确性、F1和召回率,使用2000年抽样的自举统计检验,并对分数中位数的差异进行显著性检验。使用错误发现率(FDR)方法校正产生的P值。使用两个样本(P-NET分数和密集分数)具有相同平均(预期)值的无效假设的平均值的t检验,比较在多个样本量上训练和测试P-NET和密集模型产生的五倍交叉验证的AUC,假设总体具有相同的平均(预期)值差异。同样的测试也适用于其他分数,包括recall、准确度、AUPRC、F1和准确度。对于生存分析(图2d),使用非参数对数秩检验比较两组在每个观察事件时间的危险函数估计值。平均数 t t t检验用于比较MDM4缺失后前列腺癌细胞增殖的减少与阴性对照组相比。采用卡方检验和Yates校正比较两组(原发性和转移性肿瘤患者)MDM4高扩增的预期和观察频率。
4 - 基因组规模ORF筛选分析
先前在LNCaP细胞中进行了基因组规模的ORF筛查42。简言之,用汇集的ORF文库感染细胞,经嘌呤霉素选择以分离含有相应ORF的细胞,然后用恩扎鲁胺接种在低雄激素培养基(CSS)中。在培养25天后确定每个ORF对细胞增殖的相对影响,并用Z分数表示。ORF筛选的原始结果来自Hwang等人的来源研究。我们假设P-NET识别的扩增基因调节转移性CRPC的致癌功能。为了验证这一假设,我们分析了之前发表的在LNCaP细胞中进行的基因组规模ORF筛查,该筛查确定了当过表达时,促进对AR抑制剂enzalutamide42耐药性的基因(图4b)。LNCaP细胞依赖AR,用恩扎鲁胺治疗可减弱细胞增殖。在此分析的基础上,MDM4相对于其他hits(包括细胞周期调节因子(CDK4和CDK6)或在FGF信号传导中起作用的基因(FGFR2、FGFR3和FGF6),被认为是一个强大的抗恩扎鲁胺基因;这两条途径与临床前列腺癌患者对抗雄激素治疗产生耐药性27,53有关。
5 - 对RO-5963的敏感性
LNCaP、LNCaP Abl、LNCaP 95、DU145、LAPC-4、LNCaP耐恩扎鲁胺、C4-2和PC3细胞以每孔3000个细胞的密度接种在96孔板中。24小时后,用浓度增加的RO-5963处理细胞4天。使用细胞滴度Glo测定法测定细胞增殖。使用GraphPad棱镜测定IC50值。数据以三次重复的平均值±标准差表示。实验重复三次(补充数据4中的原始数据和分析文件)。所有细胞系支原体污染检测均为阴性。对于所有公开可用的细胞系,使用STR图谱和/或直接从ATCC获得的数据进行验证
6 - MDM4基因缺失实验
在150μg ml–1 blasticin(Thermo Fisher Scientific,NC9016621)中培养耐blasticin的Cas9阳性前列腺癌细胞72小时,以富集具有最佳Cas9活性的细胞。将100万个细胞平行接种在12孔板中,并感染慢病毒,慢病毒表达针对MDM4或GFP对照的嘌呤霉素抗性sgRNAs。然后对细胞进行嘌呤霉素选择3天,然后使用Vi细胞对细胞进行计数,并进行增殖试验。7天后,用Vi细胞再次计数细胞以评估生存能力,总共12天。The target sequence against GFP was CACCGGCCACAAGTTCAGCGTGTCG (sgGFP). The target sequences against MDM4 were AGATGTTGAACACTGAGCAG (sgMDM4-1) and CTCTCCTGGACAAATCAATC (sgMDM4-2)。
7 - 免疫印迹法 8 - MDM4基因缺失降低前列腺癌细胞活力 9 - MDM4的化学抑制降低前列腺癌细胞活力
上面三个部分由于不是学生物的,就不放了,
附录
扩展数据图1
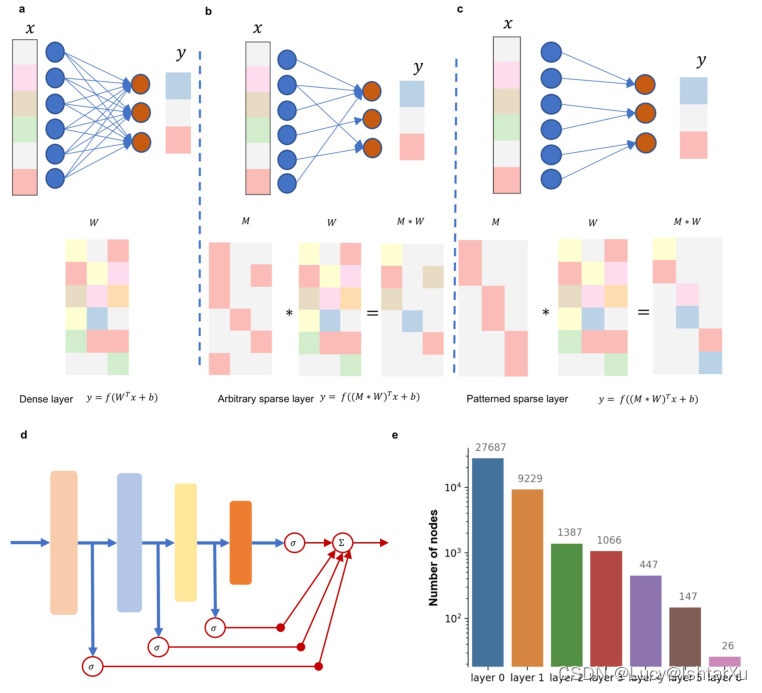
扩展数据图1 | P-NET体系结构和特征。
a、 输入为xr的稠密层∈ dx和y-R输出∈ dy向量。矩阵W-R∈ dx dy是一个可训练的权重矩阵和br∈ dy是偏置矢量。f是层激活函数。
b、 任意稀疏层都可以灵活地使用添加的M对任何连接方案进行编码∈ {0,1}dx dy⁎二元掩码矩阵,控制层的连通性,在权重矩阵上施加稀疏性。
c、 一种带遮罩矩阵M的稀疏矩阵,它遵循一定的模式。此模式可用于提高计算效率。
d、 预测节点连接到P-NET中的每一个隐层,并通过取网络中所有预测元素的平均值来计算最终预测。e、 每层P-NET的参数数。















 558
558











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









