






1、包的安装和加载
# Splatter can be installed from Bioconductor:
BiocManager::install("splatter")
# To install the most recent development version from Github use:
BiocManager::install("Oshlack/splatter", dependencies = TRUE,
+ build_vignettes = TRUE)
library("splatter")
#> Warning: package 'splatter' was built under R version 3.6.2
library("scater")
#> Warning: package 'ggplot2' was built under R version 3.6.2
library("ggplot2")2、The base Splat model
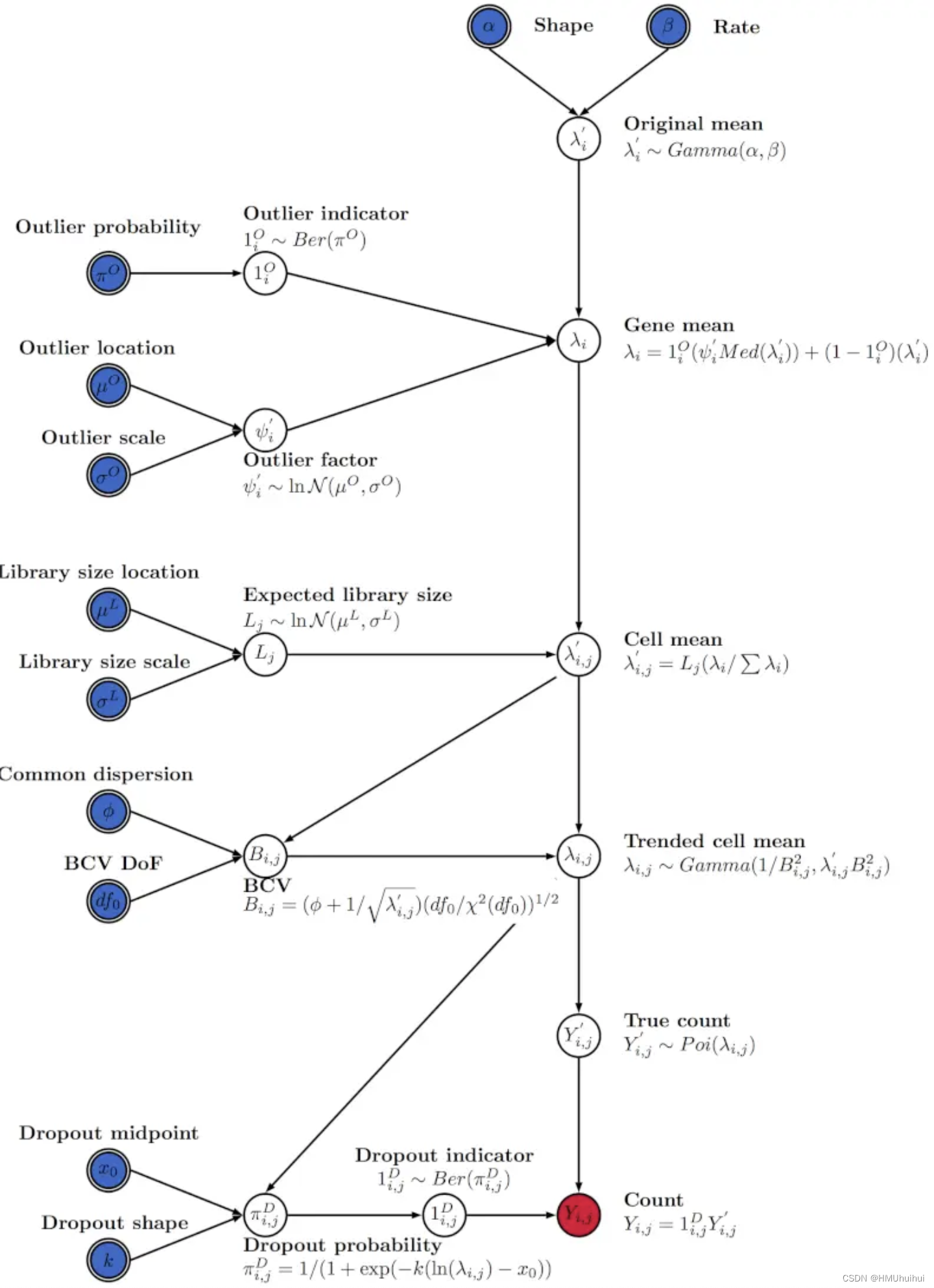
下图为Splat模型模拟的核心
3、原理及步骤
Splat模拟使用分层概率的方法,从适当的统计分布生成不同类型的数据集。
Step1:
该阶段产生每个基因的平均表达水平。(最初是从Gamma分布中选择的)对于一些被选为高表达量的异常值的基因,从对数正态分布中产生一个因子,再将这些因子乘以基因平均数的中位数,从而为这些基因创造新的均值。
Step2:该阶段包括每个细胞计数的变化。从对数正态分布中为每个细胞选择预期的库大小(总计数)。然后使用文库大小来缩放每个细胞的基因均值,从而得到模拟数据集中每个细胞的计数范围a。然后进一步调整基因均值以加强平均表达水平和变异性之间的关系。
Step3:
最终的基因表达矩阵通过使用泊松分布来生成计数矩阵,结果是由伽马-泊松(或负二项)分布的计数组成的合成数据集。附加的可选步骤来可以产生“dropout”效应:使用基于基础平均表达式水平的逻辑函数来生成“dropout”概率,然后使用伯努利分布来创建丢弃矩阵,则该矩阵将一些生成的计数设置为零。
以上描述的模型将生成单个细胞群体,但Splat模拟被设计为尽可能灵活,可以创建包括多组细胞(细胞类型)、细胞类型之间的连续路径和多个实验批次的场景。用于创建这些类型的模拟实验参数以及模型的交互如下所述:
3.1 Splat simulation parameters
在Splatter中,Splat模拟模型的参数保存在SplatParams对象中。让我们创建其中一个对象,看看它是什么样子。
params <- newSplatParams()
params
#> A Params object of class SplatParams
#> Parameters can be (estimable) or [not estimable], 'Default' or 'NOT DEFAULT'
#> Secondary parameters are usually set during simulation
#>
#> Global:
#> (Genes) (Cells) [Seed]
#> 10000 100 712777
#>
#> 28 additional parameters
#>
#> Batches:
#> [Batches] [Batch Cells] [Location] [Scale]
#> 1 100 0.1 0.1
#>
#> Mean:
#> (Rate) (Shape)
#> 0.3 0.6
#>
#> Library size:
#> (Location) (Scale) (Norm)
#> 11 0.2 FALSE
#>
#> Exprs outliers:
#> (Probability) (Location) (Scale)
#> 0.05 4 0.5
#>
#> Groups:
#> [Groups] [Group Probs]
#> 1 1
#>
#> Diff expr:
#> [Probability] [Down Prob] [Location] [Scale]
#> 0.1 0.5 0.1 0.4
#>
#> BCV:
#> (Common Disp) (DoF)
#> 0.1 60
#>
#> Dropout:
#> [Type] (Midpoint) (Shape)
#> none 0 -1
#>
#> Paths:
#> [From] [Steps] [Skew] [Non-linear] [Sigma Factor]
#> 0 100 0.5 0.1 0.8 与Splatter中的所有参数对象一样,此对象显示此模拟所需的所有参数。因为我们没有设置任何参数,所以会显示默认值,但是如果我们要更改其中的任何参数,它们都会高亮显示。我们还可以看到哪些参数可以通过Splat估计程序进行估计,哪些不可以。默认值已被选择为相当真实,但建议使用估计值来获得更接近您感兴趣的数据的模拟。可以通过在SplatParams对象中设置参数或将其直接提供给模拟函数来修改参数。
本文的其余部分提供所有这些参数的详细信息,并解释如何通过示例使用它们。
3.2 Global parameters
全局参数在每个模型中使用,并控制产生的数据集的全局特征。
# Set the number of genes to 1000
params <- setParam(params, "nGenes", 1000)
sim <- splatSimulate(params, verbose = FALSE)
dim(sim)
#> [1] 1000 1003.2.1 nCells - Number of cells
模拟的基因数量。在Splat模拟中,这不能直接设置,但必须使用batchCells参数进行控制。
3.2.2 seed - Random seed
用于生成随机数的种子,包括从分布中选择值。通过更改此值,可以生成具有相同参数的多个模拟数据集。使用相同的参数集和随机种子产生的模拟应该是相同的,但操作系统、软件版本等之间可能存在差异。
3.3 Batch parameters
通过这些参数来控制模拟数据集中的实验批次。批次效应如何包含在模型中的总体效果类似于技术重复(即多次测序相同的生物样本)。这意味着批次之间的底层结构是一致的,但是添加全局技术签名(global technical signature)可能会将它们分开。
3.3.1 Batches - Number of batches
模拟中的批次数。不能直接设置,但可以通过设batchCells来进行控制。
3.3.2 batchCells - Cells per batch
指定每批中的细胞数量的向量。批次数(nBatches)等于向量的长度,细胞数(nCells)等于总和。
# Simulation with two batches of 100 cells
sim <- splatSimulate(params, batchCells = c(100, 100), verbose = FALSE)
# PCA plot using scater
sim <- logNormCounts(sim)
sim <- runPCA(sim)
plotPCA(sim, colour_by = "Batch")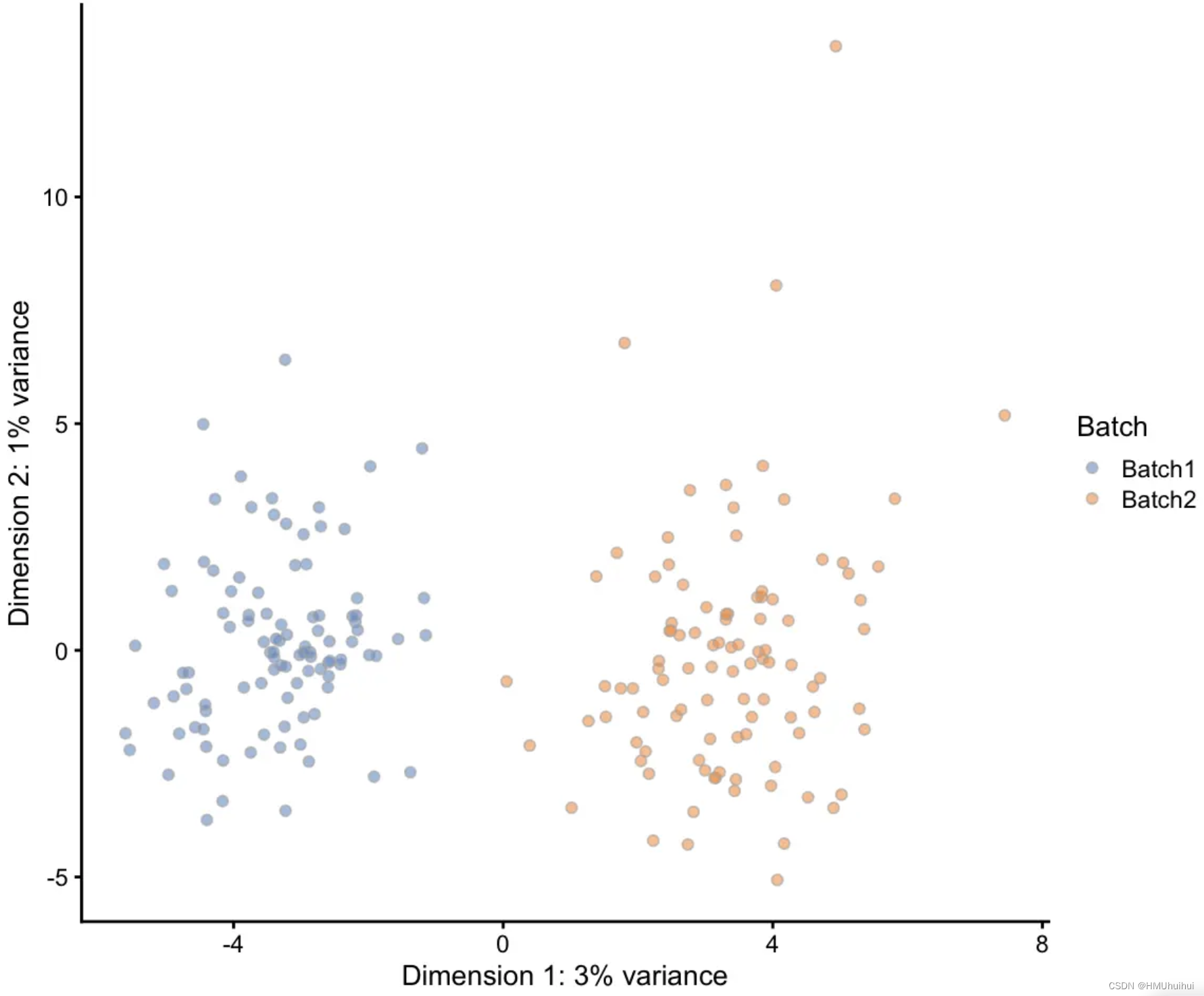
3.3.3 batch.facLoc - Batch factor location and batch.facScale - Batch factor scale
通过从对数正态分布为每个批次中的每个基因生成一个小比例因子来指定批次,然后将这些因子应用于每个批次中的潜在基因,修改这些参数生成更大或更小的因子来影响批次之间的差异程度。
# Simulation with small batch effects
sim1 <- splatSimulate(params, batchCells = c(100, 100),
batch.facLoc = 0.001, batch.facScale = 0.001,
verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Batch") + ggtitle("Small batch effects")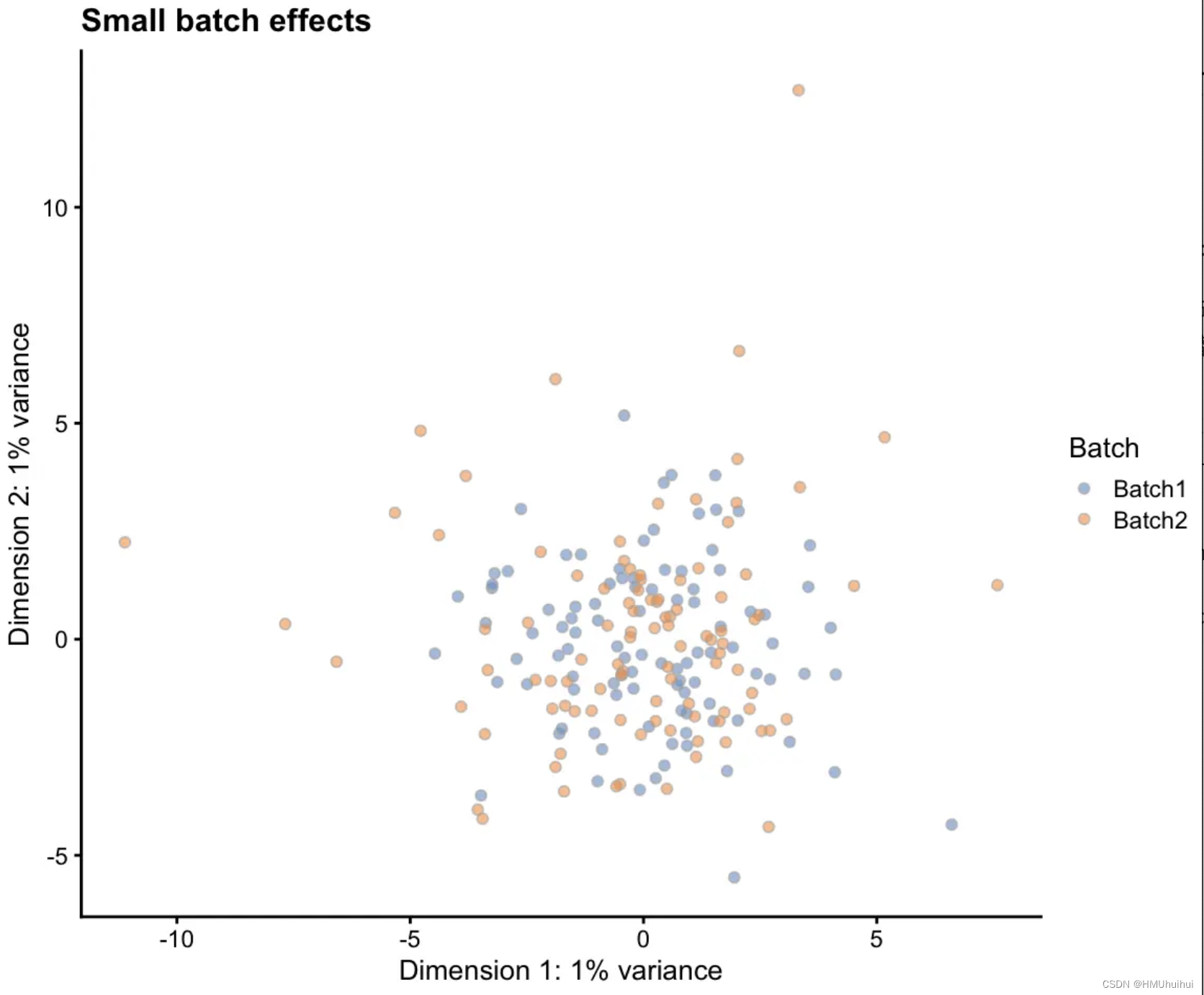
# Simulation with big batch effects
sim2 <- splatSimulate(params, batchCells = c(100, 100),
batch.facLoc = 0.5, batch.facScale = 0.5,
verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Batch") + ggtitle("Big batch effects")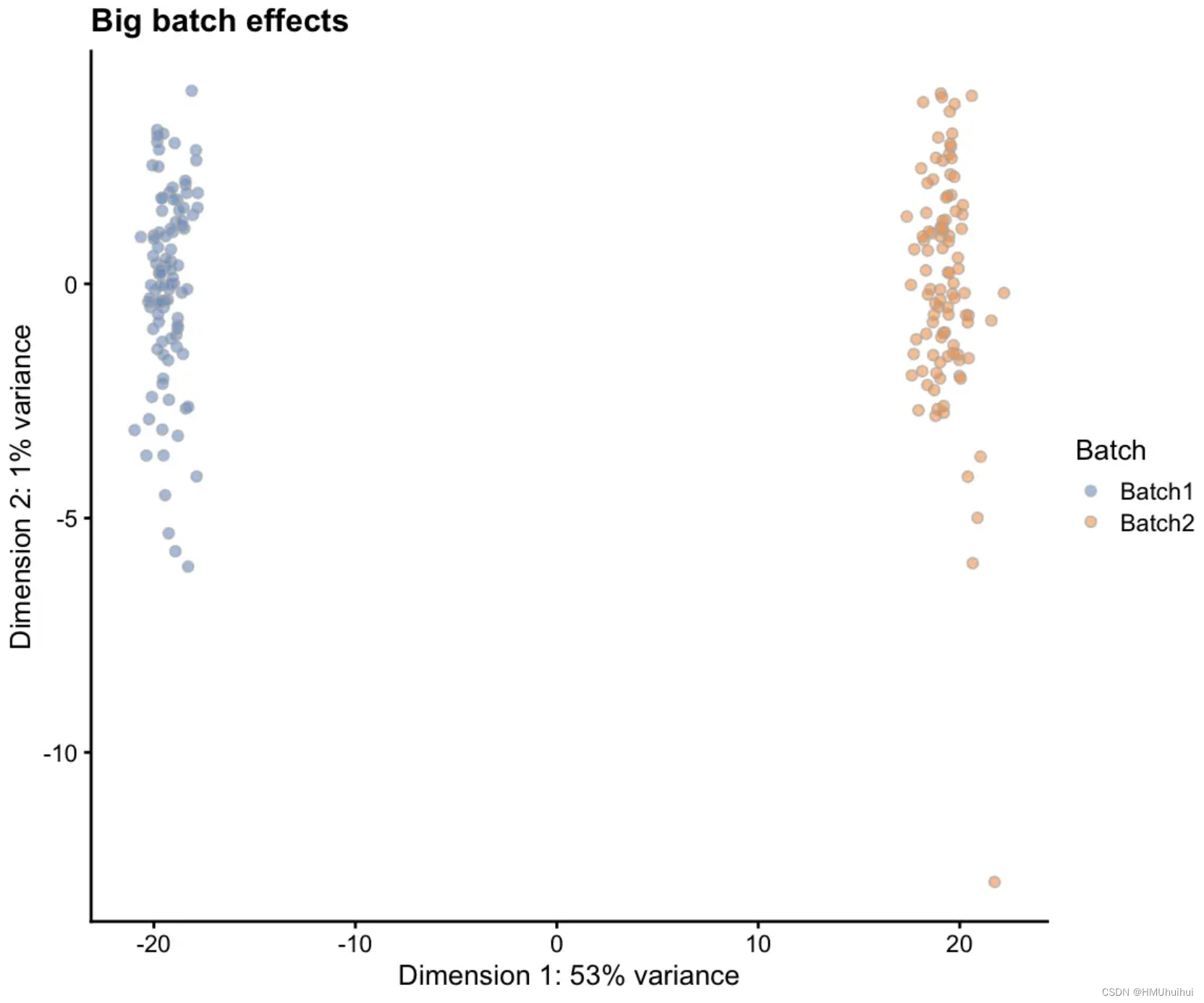
3.4 Mean parameters
通过设置这些参数来控制用于生成潜在原始基因均值的分布
3.4.1 mean.shape - Mean shape and mean.rate - Mean rate
这些参数控制提取基因平均值的伽玛分布。形状和速率之间的关系可能很复杂,使用从实际数据集中估计的值通常比尝试用手动设置它们要好。虽然这些参数控制碱基基因,但最终模拟中的方法将取决于模型的其他部分,特别是模拟的每种细胞counts总数(文库大小)。
3.5 Library size parameters
这些参数控制每个细胞的预期计数数。注意:由于采样的原因,最终模拟中每个细胞的实际计数可能不同,启用dropout效果也会对此产生影响,为了保持一致,我们在这里使用术语library size,使得预期总数更合适。
3.5.1 lib.loc - Library size location and lib.scale - Library size scale
这些参数控制用于为每个细胞生成库大小的分布形状。增加
lib.loc将导致每个细胞的计数更多,而增加lib.scale将导致每个细胞的计数更具可变性。
3.5.2 lib.norm - Library size distribution
在Splat模拟中,库大小使用的默认(和推荐)分布是对数正态分布。然而,在极少数情况下,正态分布可能更合适。将
lib.norm设置为TRUE将使用正态分布,而不是对数正态分布。
3.6 Expression outlier parameters
在开发Splat模拟时,我们发现虽然伽玛分布通常与某些数据集的基因均值匹配良好,但它没有正确捕获高表达的基因。为此,我们将表达异常值添加到Splat模型。
3.6.2 out.prob - Expression outlier probability
该参数控制基因被选为表达异常值的概率,较高的值将导致更多的异常值基因。
# Few outliers
sim1 <- splatSimulate(out.prob = 0.001, verbose = FALSE)
ggplot(as.data.frame(rowData(sim1)),
aes(x = log10(GeneMean), fill = OutlierFactor != 1)) +
geom_histogram(bins = 100) +
ggtitle("Few outliers")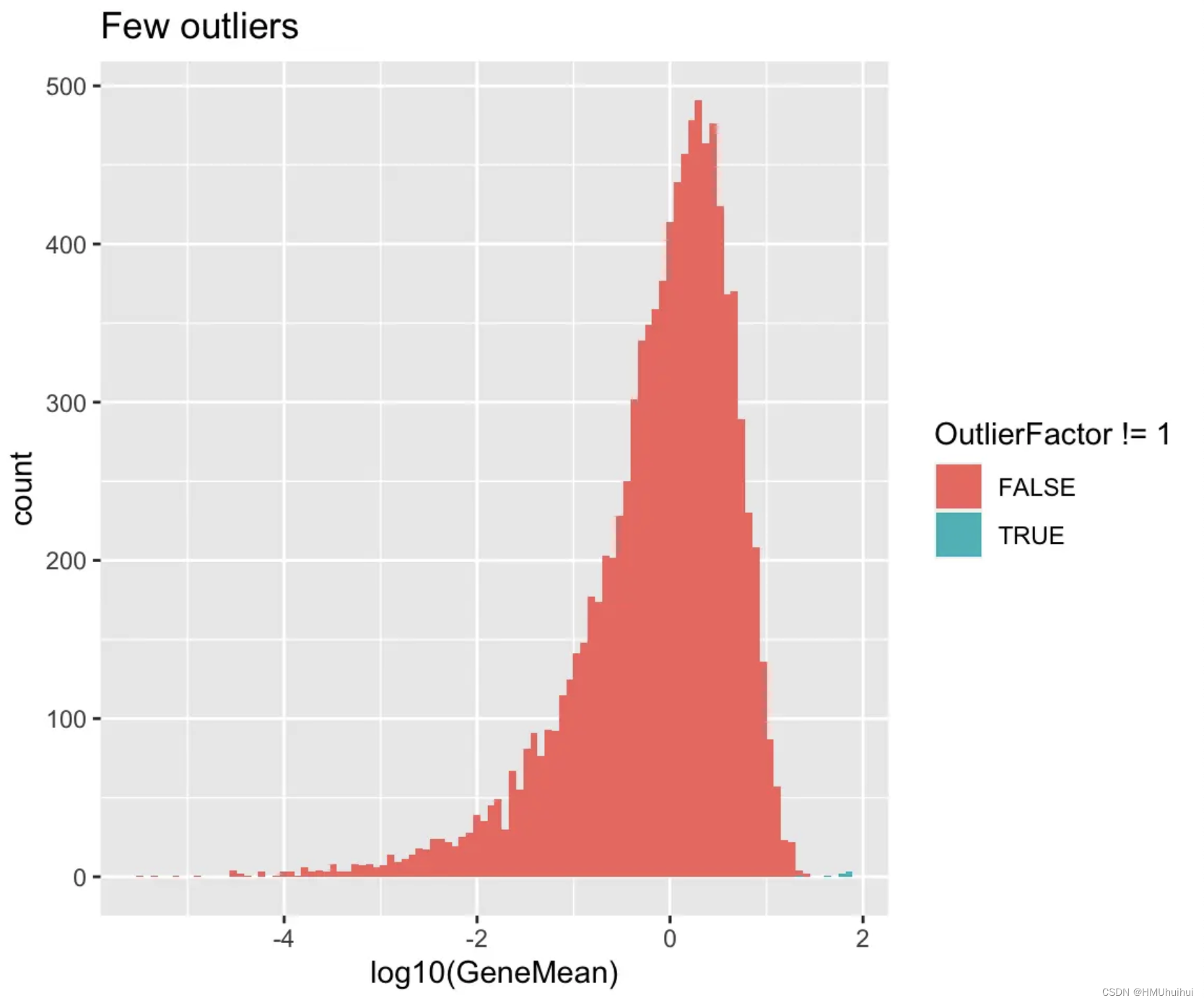
# Lots of outliers
sim2 <- splatSimulate(out.prob = 0.2, verbose = FALSE)
ggplot(as.data.frame(rowData(sim2)),
aes(x = log10(GeneMean), fill = OutlierFactor != 1)) +
geom_histogram(bins = 100) +
ggtitle("Lots of outliers")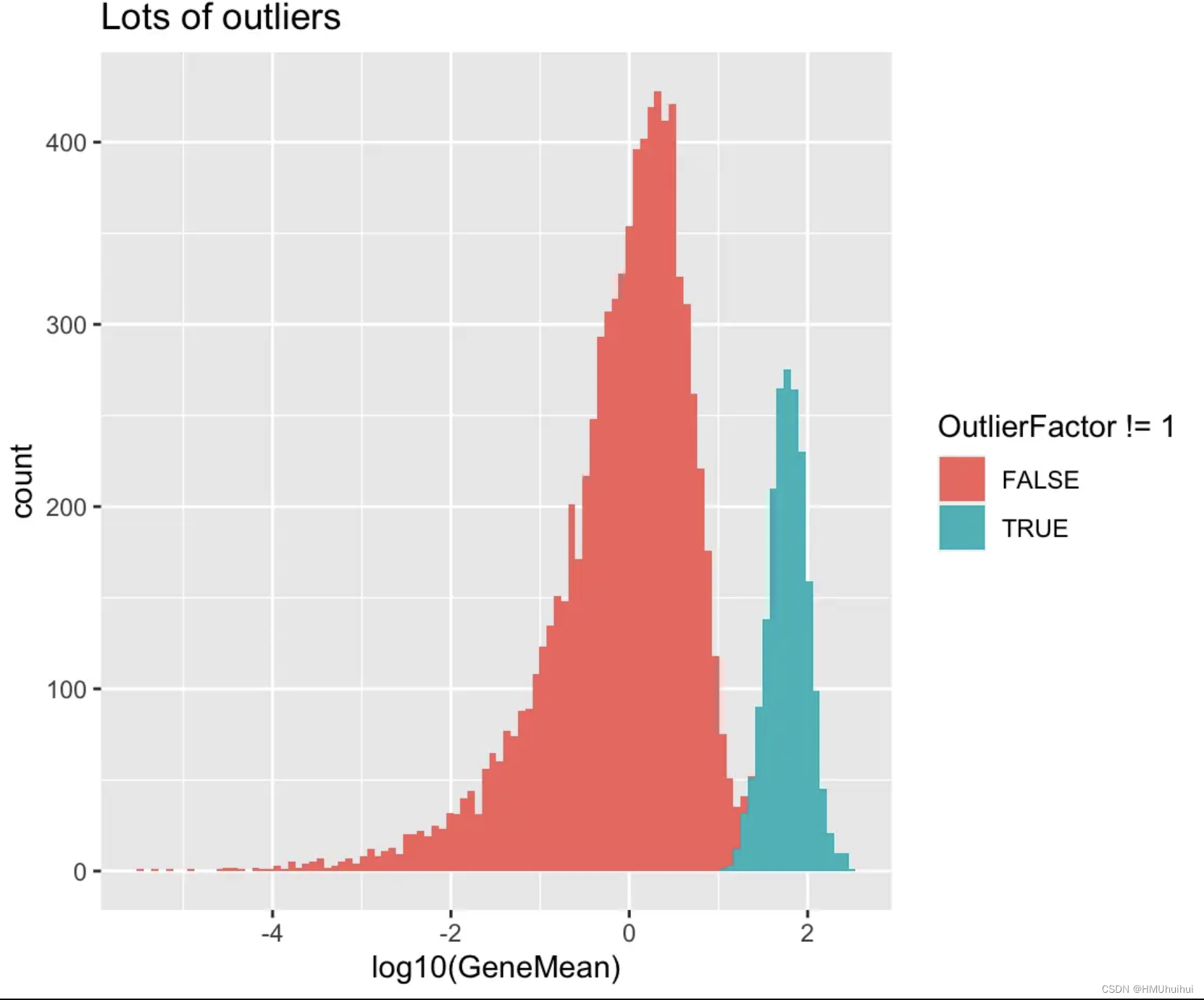
3.6.3 Group parameters
在此模拟阶段之前,只考虑了单个细胞群体,但我们通常希望模拟具有多种细胞的数据集。我们通过将细胞分配到组来做到这一点。
3.6.4 nGroups - Number of groups
要模拟的组数,此参数不能直接设置,可使用group.prob进行控制。
3.6.5 group.prob - Group probabilities
给出细胞将被分配到组的概率的向量,向量的长度给出组(nGroups)的数量,并且概率总和必须为1。调整概率的数量和相对值会更改组的数量和相对大小。要模拟分组,我们还需要使用splatSimulateGroups函数或设置method = "groups"。
params.groups <- newSplatParams(batchCells = 500, nGenes = 1000)
# One small group, one big group
sim1 <- splatSimulateGroups(params.groups, group.prob = c(0.9, 0.1),
verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Group") + ggtitle("One small group, one big group")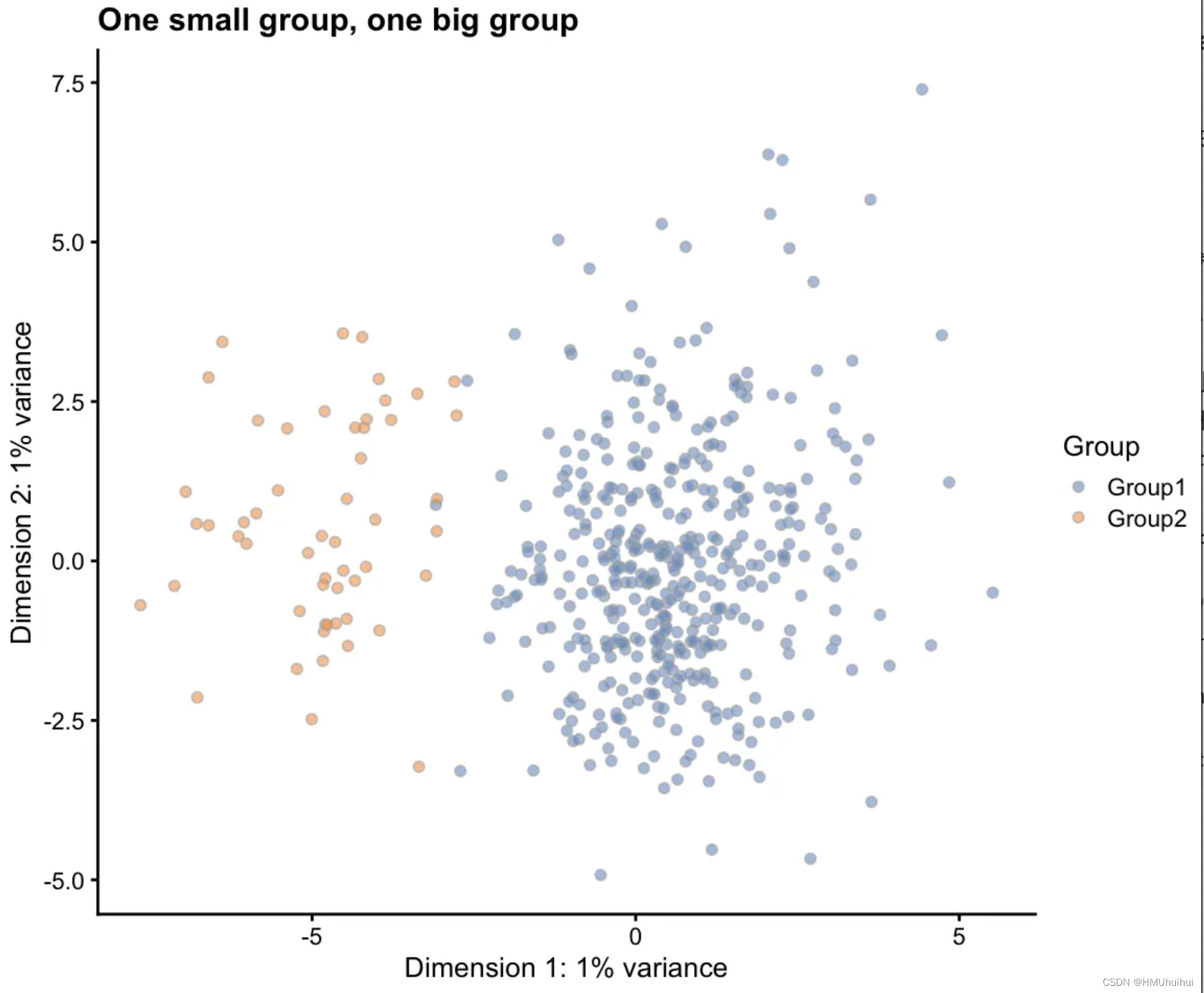
# Five groups
sim2 <- splatSimulateGroups(params.groups,
group.prob = c(0.2, 0.2, 0.2, 0.2, 0.2),
verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Group") + ggtitle("Five groups")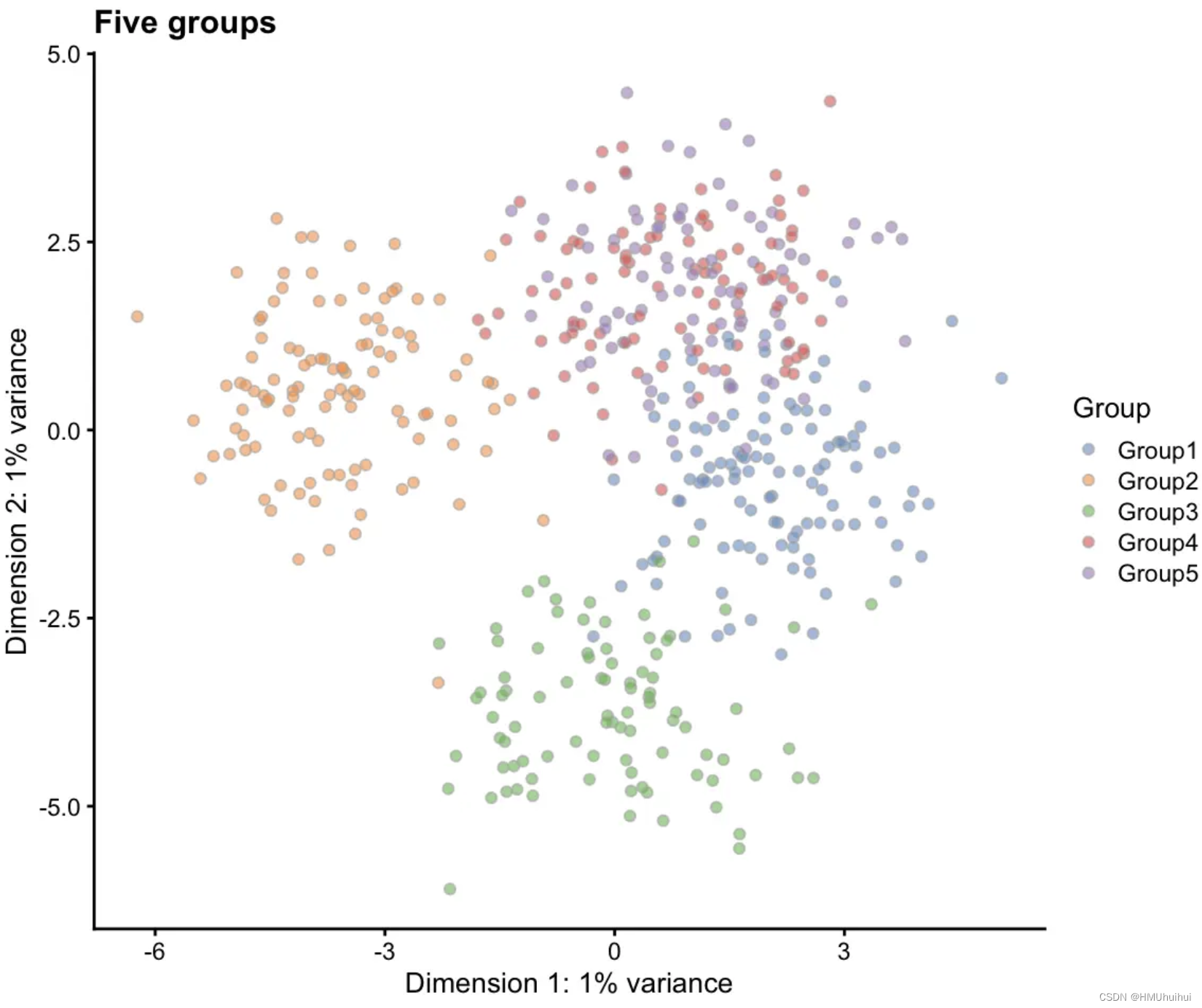
注意:一旦有三个或四个以上的组,就很难在PCA空间中正确地查看它们。为简单起见,我们在这里使用PCA,但通常非线性降维(如t-SNE或UMAP)是可视化分组的更有用的方式。
3.6.6 Differential expression parameters
通过修改所选基因的基本表达水平来创建不同的组。这样做的过程是模拟每个组和一个虚构的基底细胞之间的差异表达(DE)。改变差异表达参数可控制组之间的相似程度。
3.6.7 de.prob - DE probability
此参数控制选择基因进行差异表达的概率。
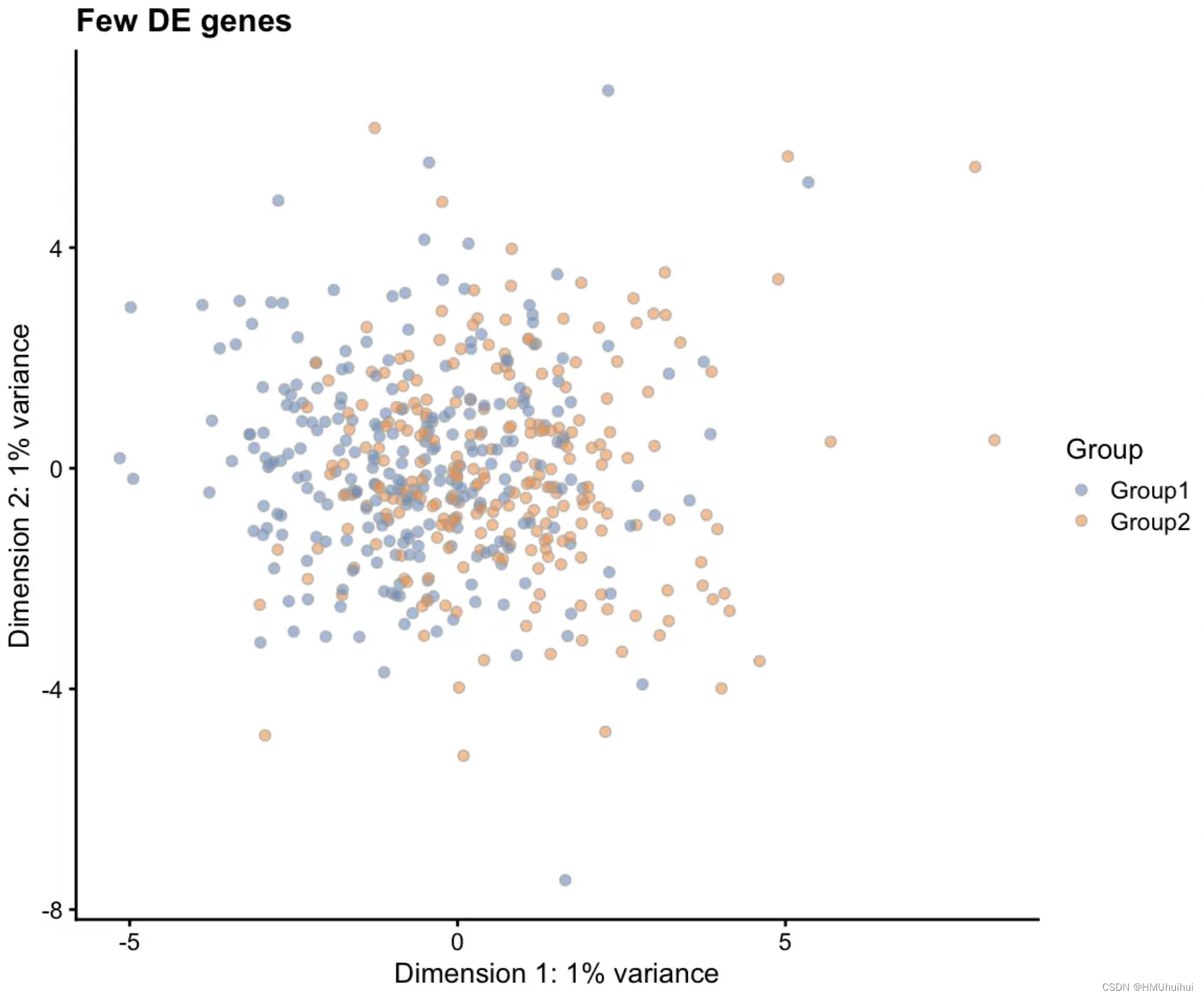
# Few DE genes
sim1 <- splatSimulateGroups(params.groups, group.prob = c(0.5, 0.5),
de.prob = 0.01, verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Group") + ggtitle("Few DE genes")
# Lots of DE genes
sim2 <- splatSimulateGroups(params.groups, group.prob = c(0.5, 0.5),
de.prob = 0.3, verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Group") + ggtitle("Lots of DE genes")
3.6.8 de.downProb - Down-regulation probability
选定的DE基因既可以下调(因子小于1),也可以上调(因子大于1)。此参数控制选定基因下调的概率。
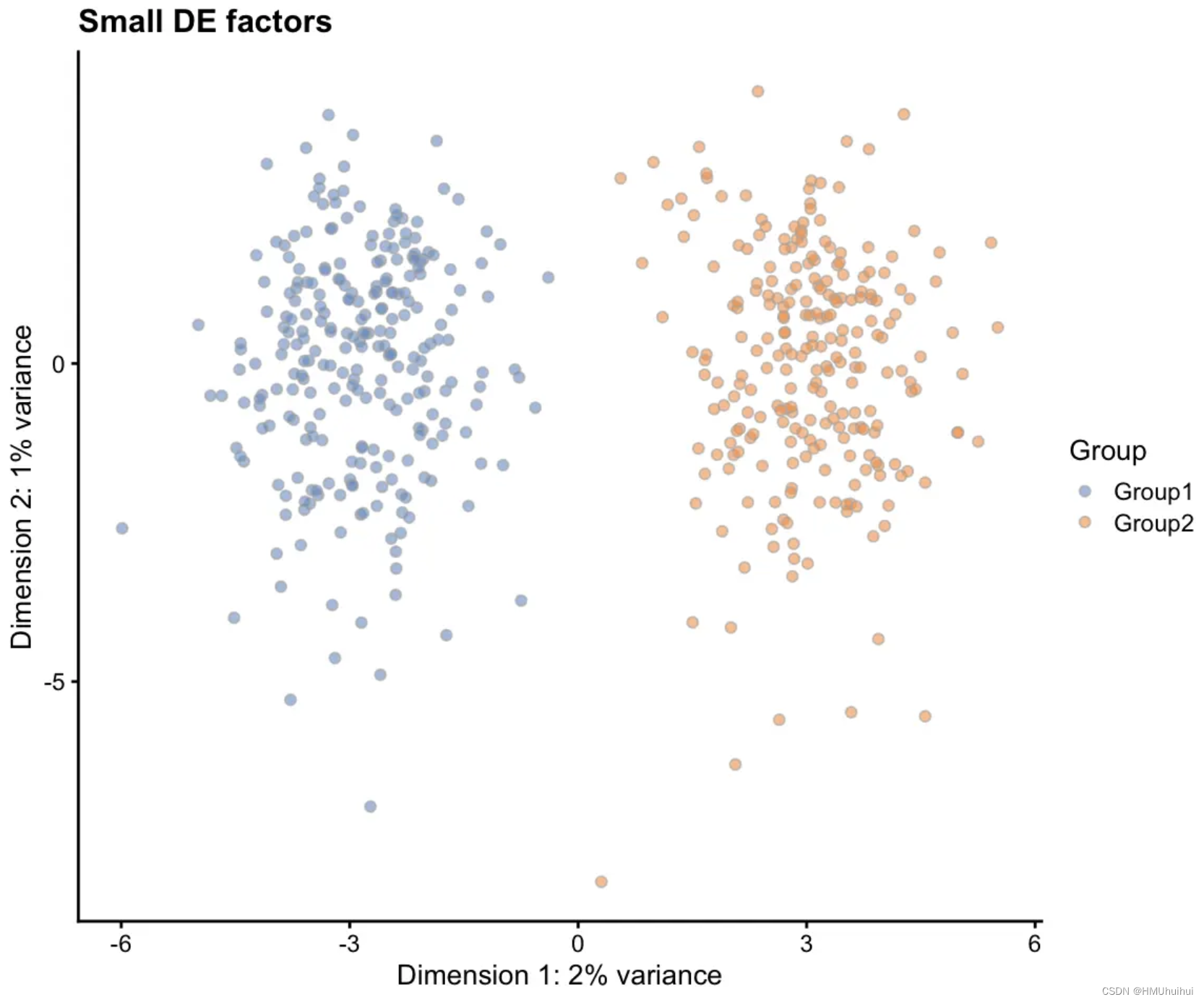
3.6.9 de.facLoc - DE factor location and de.facScale - DE factor scale
差异表达因子以类似于批次效应因子和表达异常值因子的方式从对数正态分布中产生。更改这些参数可能会导致组之间或多或少存在极端差异。
# Small DE factors
sim1 <- splatSimulateGroups(params.groups, group.prob = c(0.5, 0.5),
de.facLoc = 0.01, verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Group") + ggtitle("Small DE factors")
# Big DE factors
sim2 <- splatSimulateGroups(params.groups, group.prob = c(0.5, 0.5),
de.facLoc = 0.3, verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Group") + ggtitle("Big DE factors")
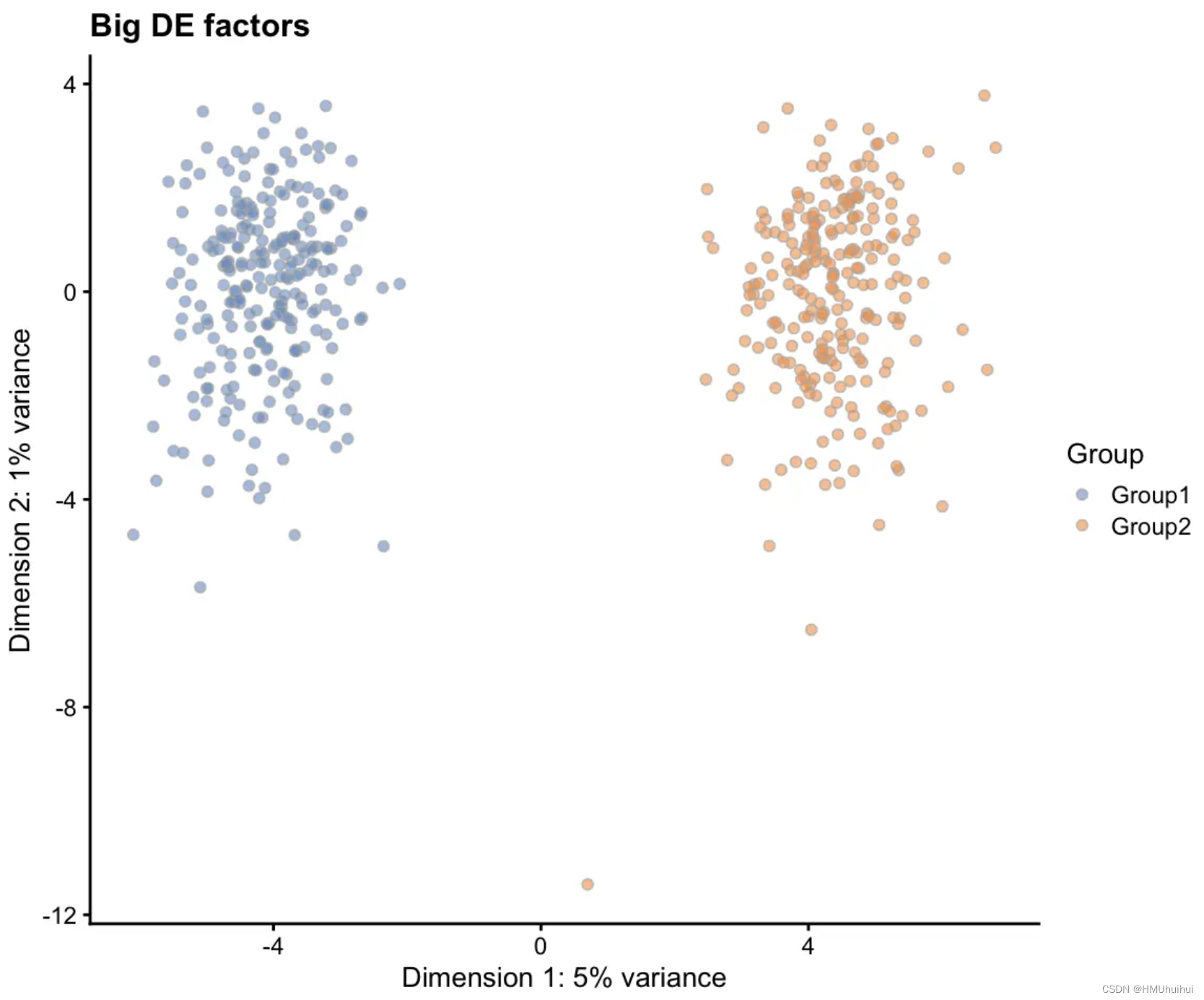
仅从PCA图来看,此效果似乎类似于调整de.prob,但效果是以不同的方式实现的。较高的de.prob意味着更多的基因有差异表达,但是改变DE因子会改变相同数量基因的DE水平。
3.6.10 Complex differential expression
通过提供值向量,可以为每组指定每个差异表达参数。这些向量的长度必须与group.prob相同。将参数指定为向量允许进行更复杂的模拟,其中组间或多或少差异,而不是同等差异。以下是一些不同DE场景的示例。
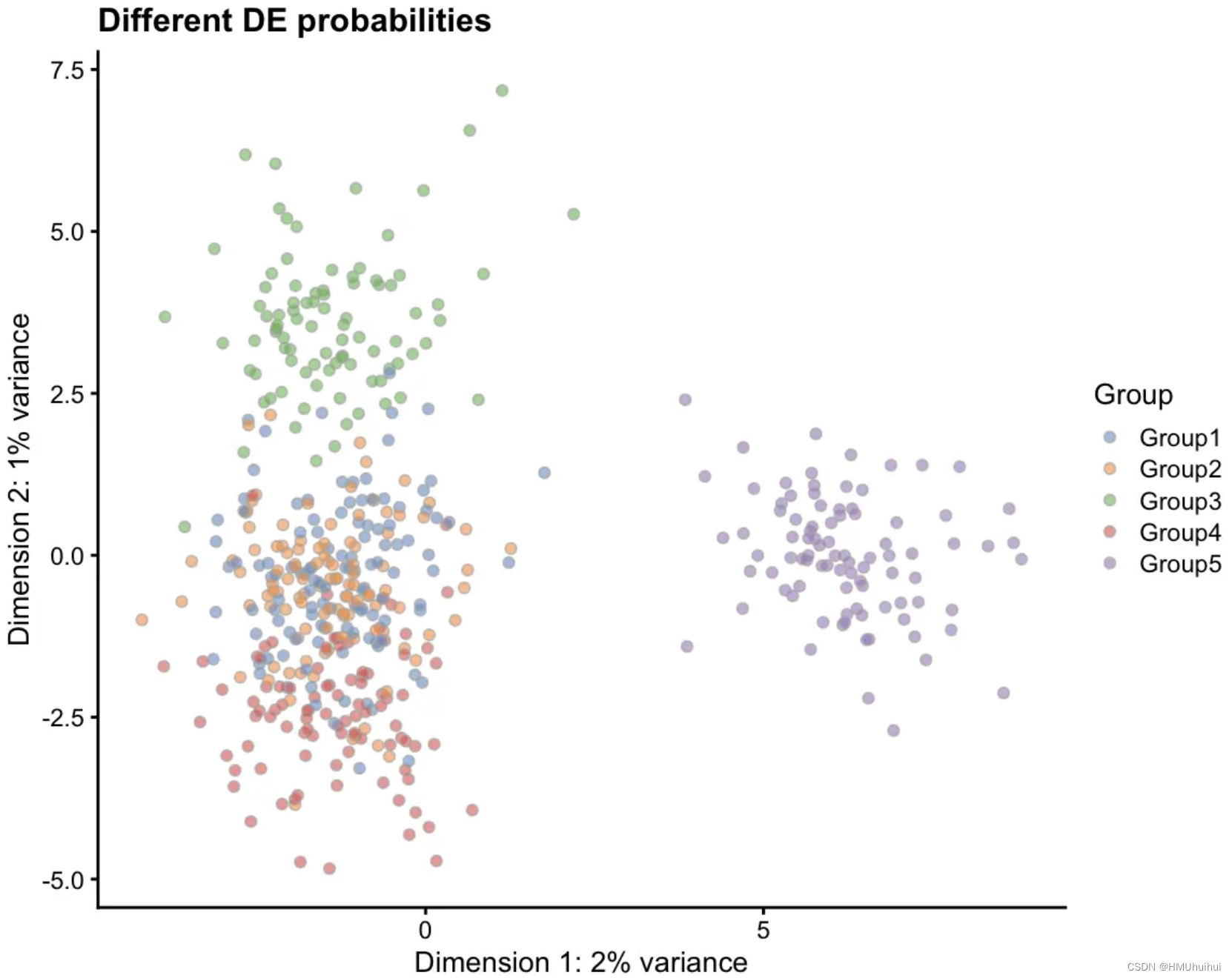
# Different DE probs
sim1 <- splatSimulateGroups(params.groups,
group.prob = c(0.2, 0.2, 0.2, 0.2, 0.2),
de.prob = c(0.01, 0.01, 0.1, 0.1, 0.3),
verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
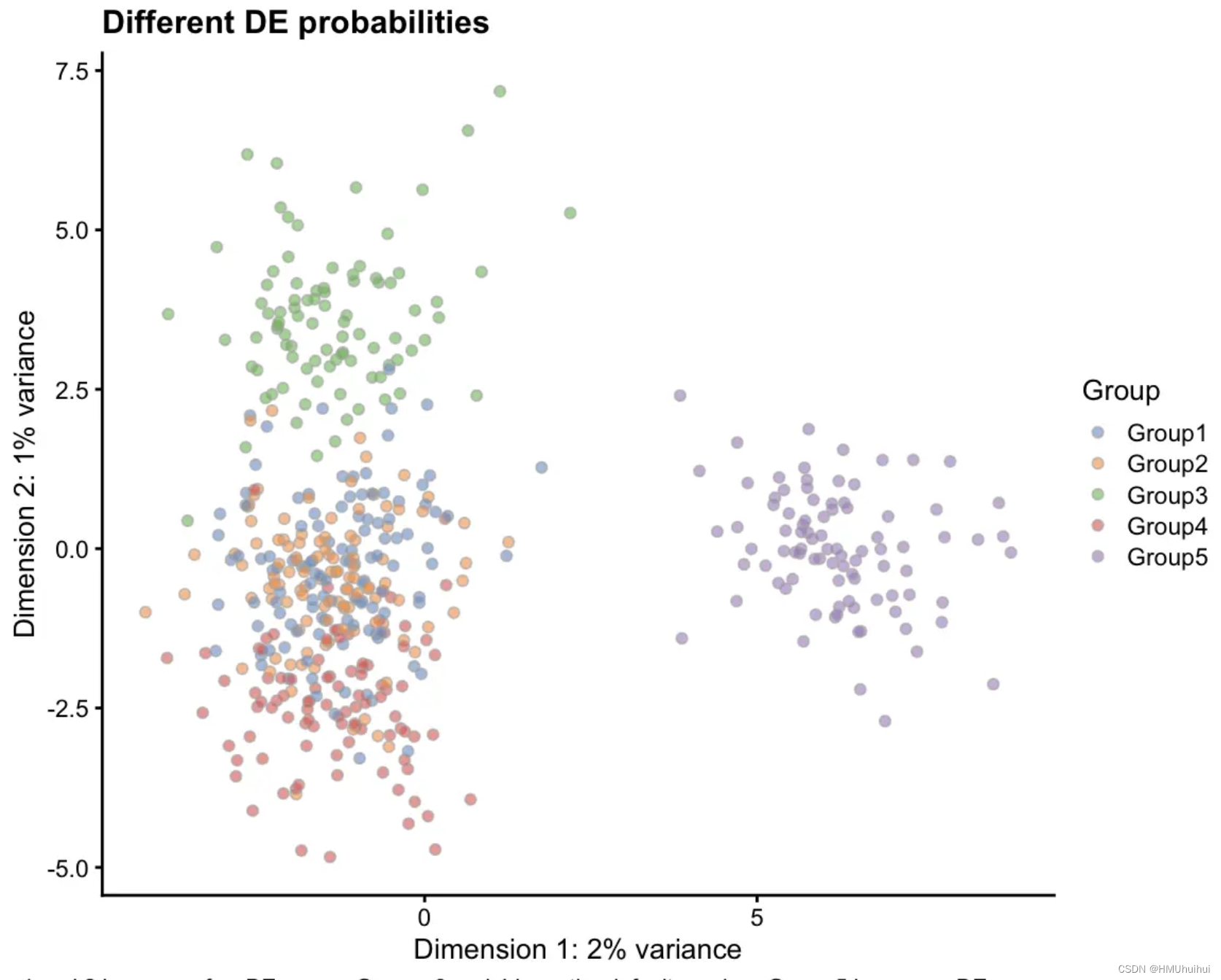
plotPCA(sim1, colour_by = "Group") +
labs(title = "Different DE probabilities",
caption = paste("Groups 1 and 2 have very few DE genes,",
"Groups 3 and 4 have the default number,",
"Group 5 has many DE genes"))
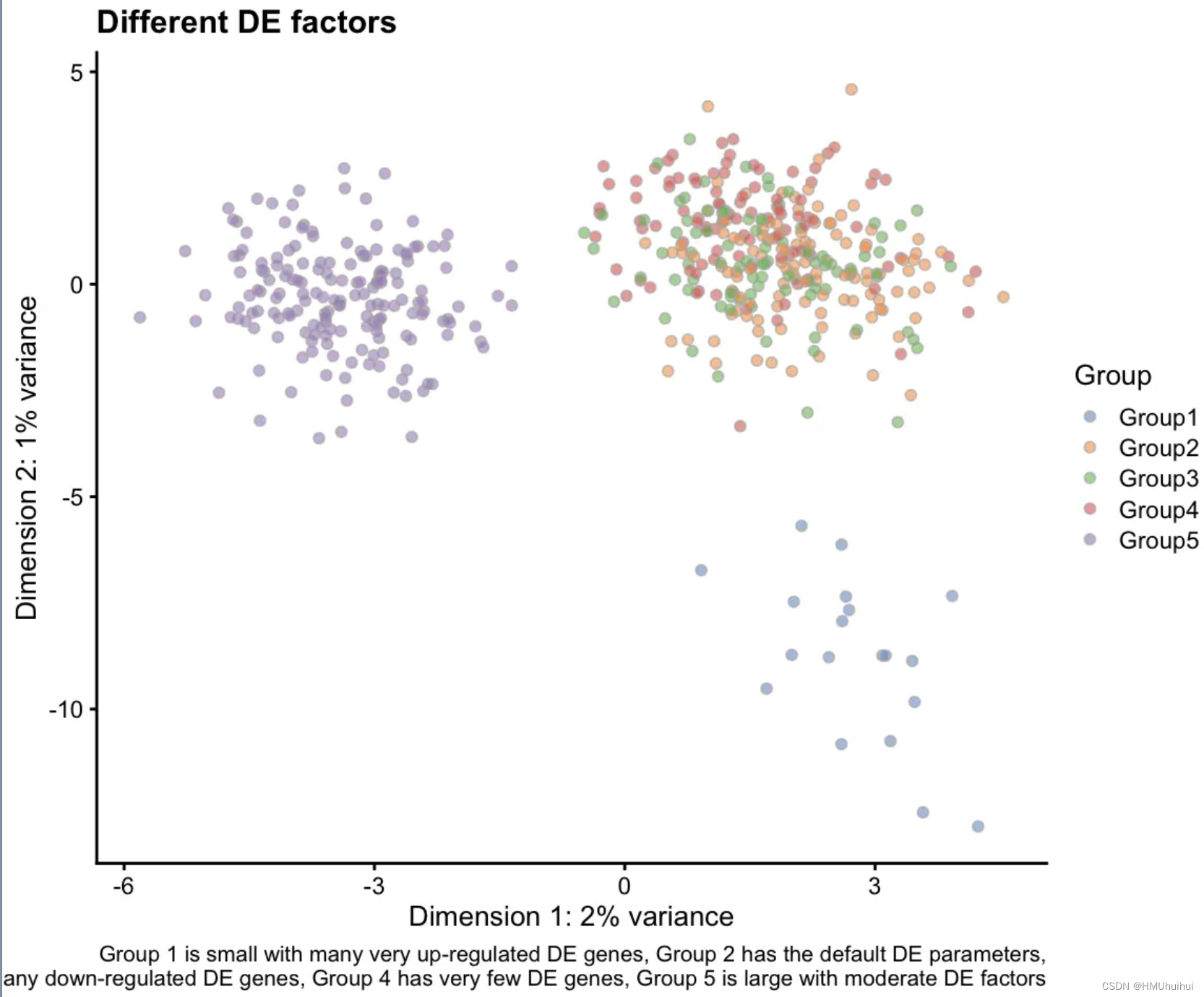
# Different DE factors
sim2 <- splatSimulateGroups(params.groups,
group.prob = c(0.2, 0.2, 0.2, 0.2, 0.2),
de.facLoc = c(0.01, 0.01, 0.1, 0.1, 0.2),
de.facScale = c(0.2, 0.5, 0.2, 0.5, 0.4),
verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Group") +
labs(title = "Different DE factors",
caption = paste("Group 1 has factors with small location (value),",
"and scale (variability),",
"Group 2 has small location and greater scale.\n",
"Groups 3 and 4 have greater location with small,",
"and large scales",
"Group 5 has bigger factors with moderate",
"variability"))
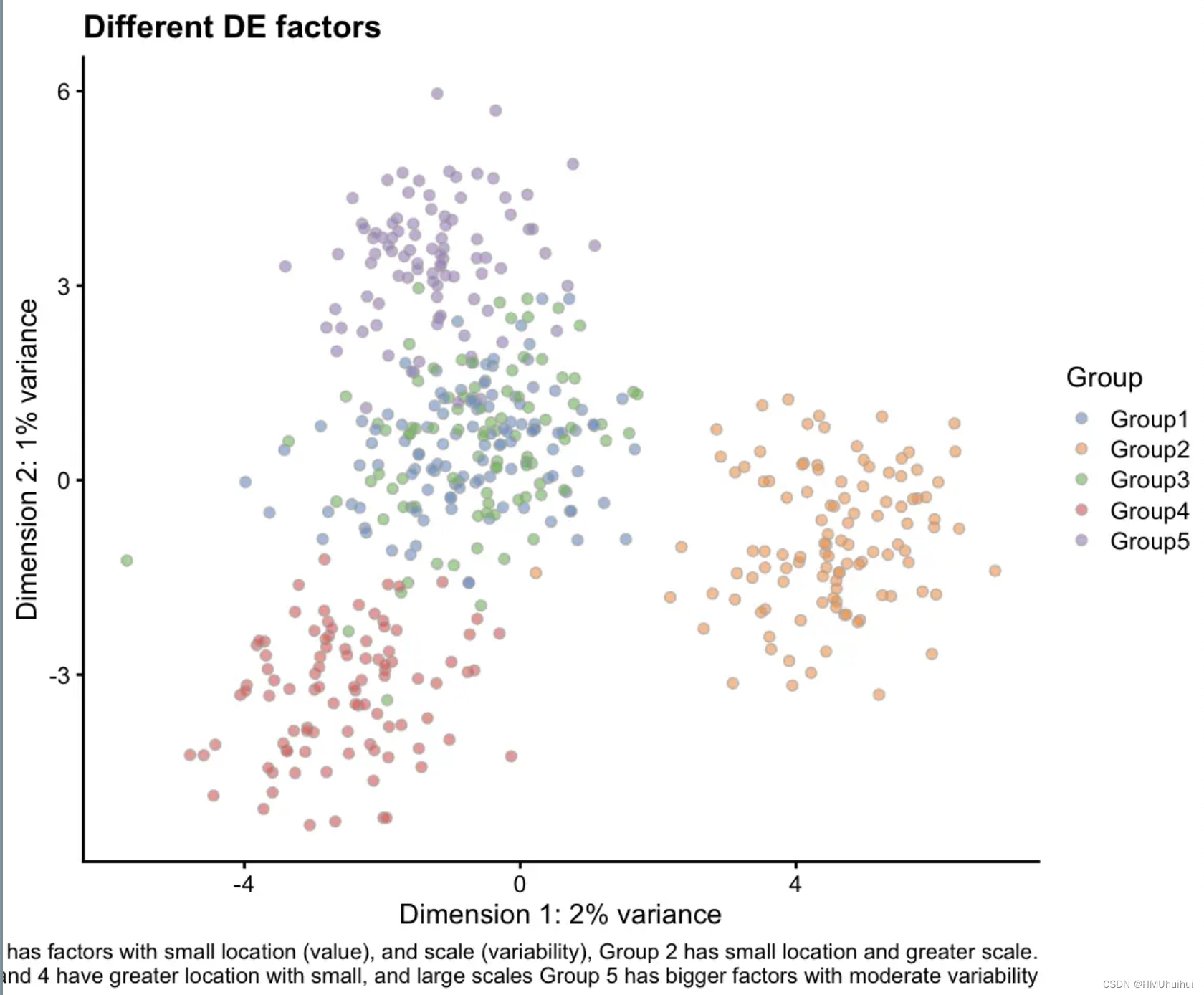
# Combination of everything
sim3 <- splatSimulateGroups(params.groups,
group.prob = c(0.05, 0.2, 0.2, 0.2, 0.35),
de.prob = c(0.3, 0.1, 0.2, 0.01, 0.1),
de.downProb = c(0.1, 0.4, 0.9, 0.6, 0.5),
de.facLoc = c(0.6, 0.1, 0.1, 0.01, 0.2),
de.facScale = c(0.1, 0.4, 0.2, 0.5, 0.4),
verbose = FALSE)
sim3 <- logNormCounts(sim3)
sim3 <- runPCA(sim3)
plotPCA(sim3, colour_by = "Group") +
labs(title = "Different DE factors",
caption = paste(
"Group 1 is small with many very up-regulated DE genes,",
"Group 2 has the default DE parameters,\n",
"Group 3 has many down-regulated DE genes,",
"Group 4 has very few DE genes,",
"Group 5 is large with moderate DE factors")
)
3.7 Biological Coefficient of Variation (BCV) parameters
BCV参数控制模拟数据集中基因的可变性。
3.7.1 bcv.common - Common BCV
公共参数bcv.common控制数据集中所有基因的潜在公共可变性。
3.7.2 bcv.df - BCV Degrees of Freedom
此参数设置BCV反卡方分布中使用的自由度。改变这一点会改变平均表达对基因变异性的影响。
3.8 Dropout parameters
这些参数控制是否添加附加丢弃以增加模拟数据集中的零数,以及是否应用该参数。
3.8.1 dropout.type - Dropout type
此参数确定要模拟的dropout效果的类型。将其设置为"none"表示不会dropout,“experiment”表示对每个细胞使用相同的参数集,"batch"对同一批次中的每个细胞使用相同的参数,"group"对同一组中的每个细胞使用相同的参数集,"cell"对每个单细胞使用不同的参数集。
3.8.2 dropout.mid - Dropout mid point and dropout.shape - Dropout shape
特定细胞中的特定计数被设置为零的概率与该基因在该细胞中的平均表达有关。该关系使用带有这些参数的逻辑函数来表示。dropout.mid参数控制概率等于0.5的点,dropout.shape控制概率如何随表达式的增加而变化。这些参数必须是具有适当长度的向量,具体取决于所选的丢弃类型(dropout type)。
3.9Path parameters
对于许多用途,模拟分组就足够了,但在某些情况下,模拟细胞类型之间的连续变化更合适。路径的数量和将细胞分配给它们的概率仍然由group.prob参数控制,沿路径的变化量由DE参数控制,但是splatSimulatePath模型的其他方面由这些参数控制。
3.9.1 path.from - Path origin
此参数控制差异化路径的顺序。它是一个与group.prob长度相同的向量,提供了每条路径的起始位置。例如,c(0,1,1,3)的path.from将指示路径1从原点(0)开始,路径2从路径1的末端开始,路径3也从路径1(分支点)的末端开始,路径4从路径3的末端开始。
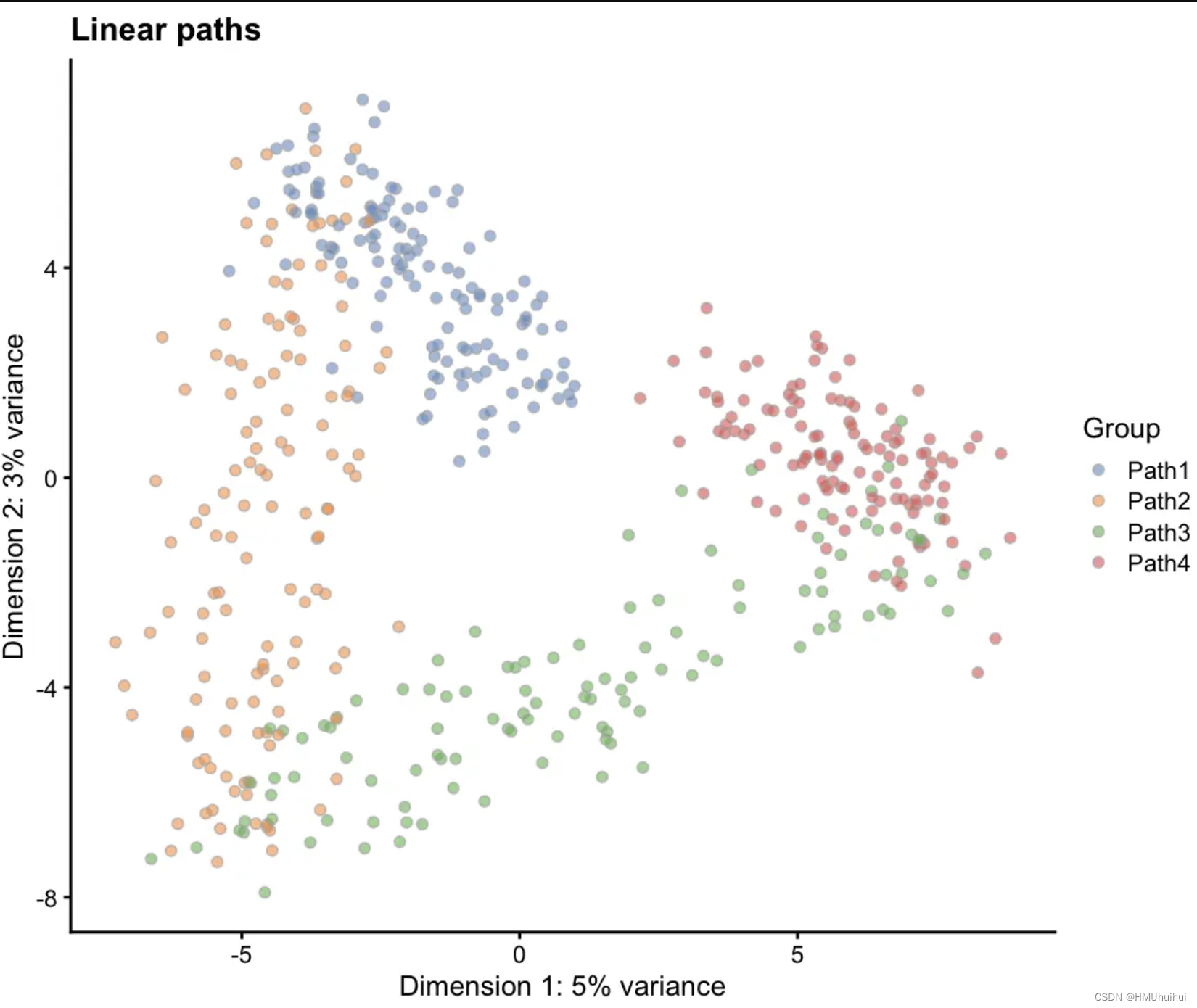
# Linear paths
sim1 <- splatSimulatePaths(params.groups,
group.prob = c(0.25, 0.25, 0.25, 0.25),
de.prob = 0.5, de.facLoc = 0.2,
path.from = c(0, 1, 2, 3),
verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Group") + ggtitle("Linear paths")
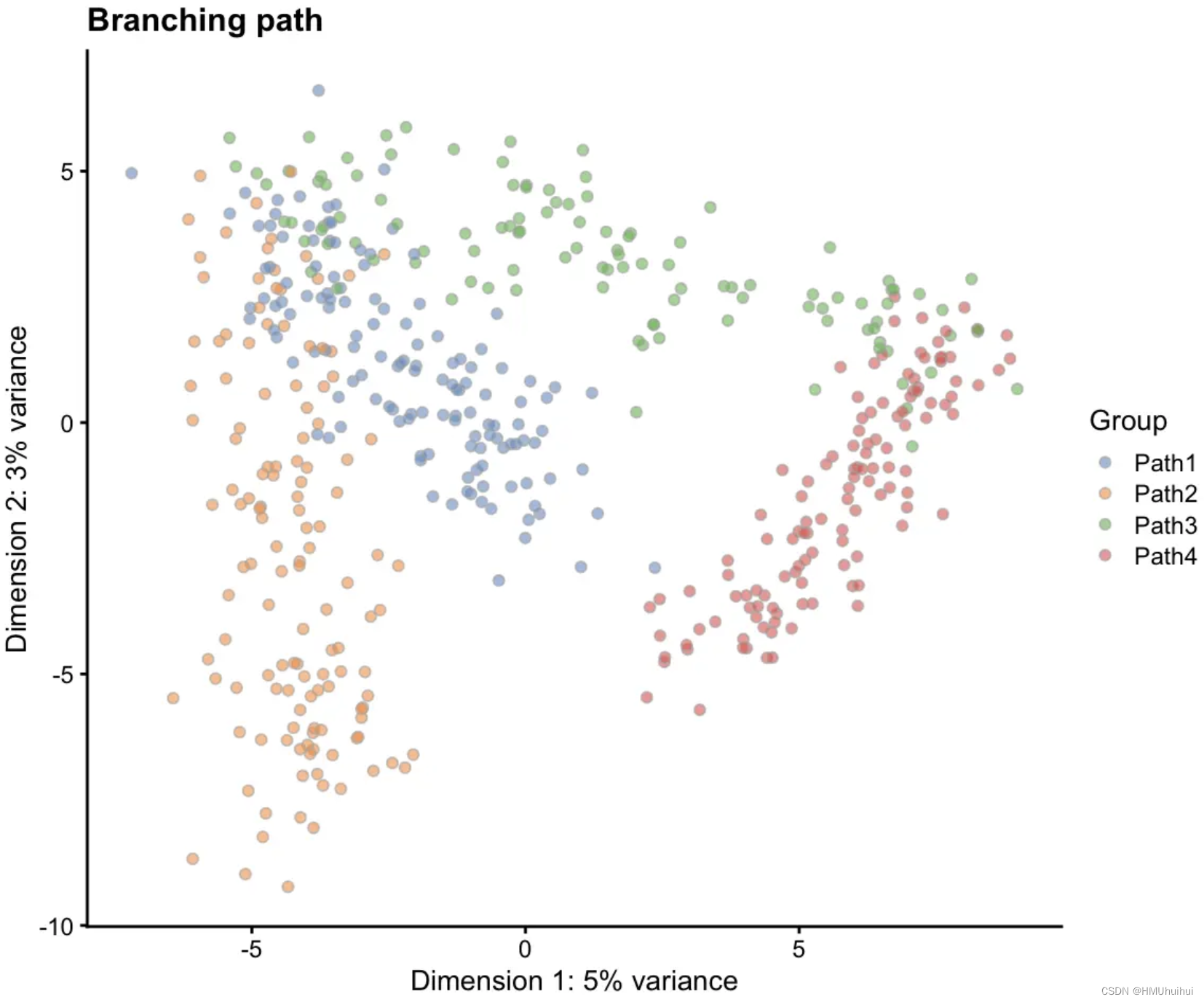
# Branching path
sim2 <- splatSimulatePaths(params.groups,
group.prob = c(0.25, 0.25, 0.25, 0.25),
de.prob = 0.5, de.facLoc = 0.2,
path.from = c(0, 1, 1, 3),
verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Group") + ggtitle("Branching path")
3.9.2 path.nSteps - Number of steps
路径是通过使用与用于组的相同的微分表达式过程来创建的,以生成端点。然后使用Interpolation在起点和终点之间创建一系列步骤。此参数控制沿路径的步数,从而控制路径的离散或平滑程度。
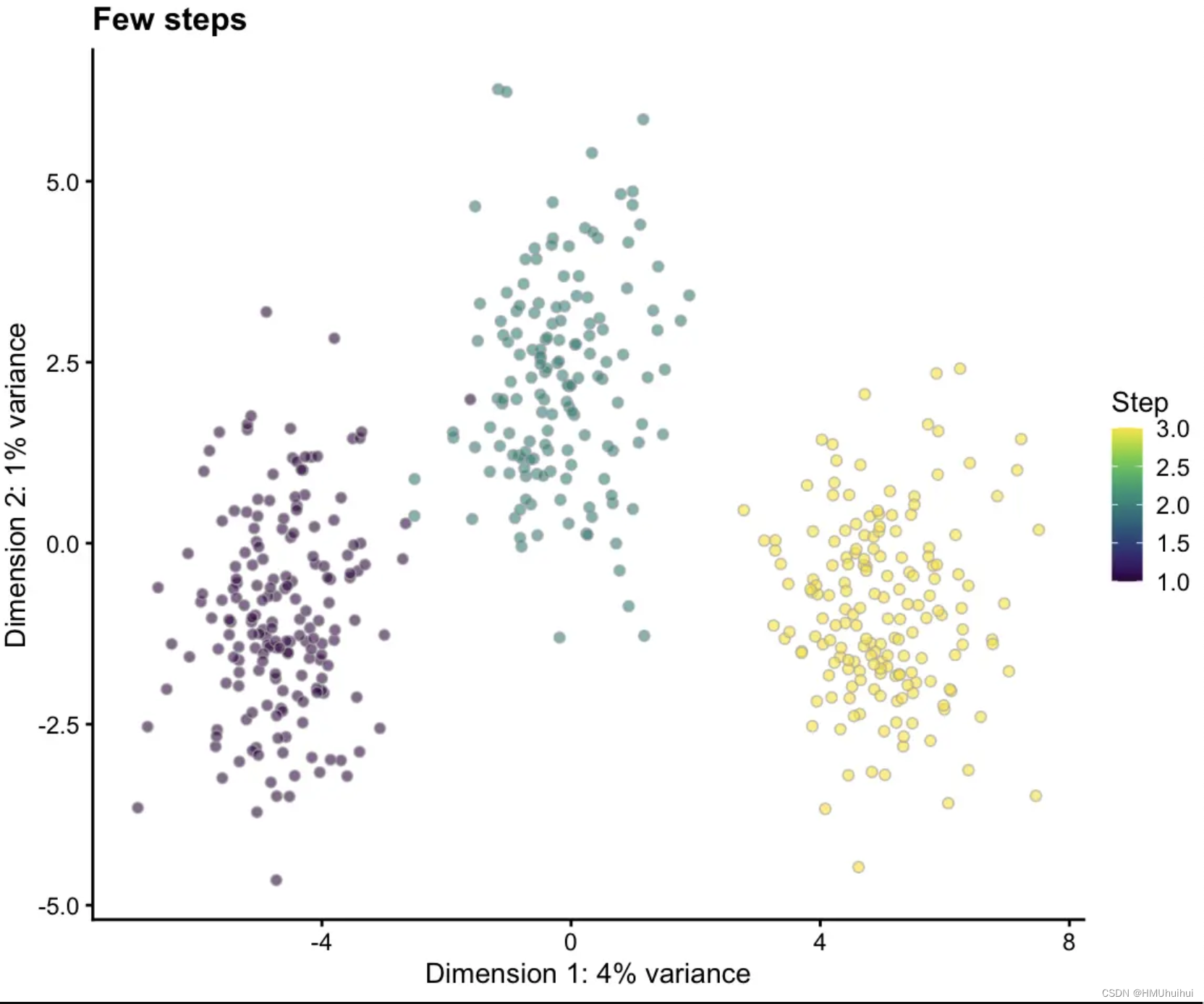
# Few steps
sim1 <- splatSimulatePaths(params.groups, path.nSteps = 3,
de.prob = 0.5, de.facLoc = 0.2, verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Step") + ggtitle("Few steps")
# Lots of steps
sim2 <- splatSimulatePaths(params.groups, path.nSteps = 1000,
de.prob = 0.5, de.facLoc = 0.2, verbose = FALSE)
sim2 <- logNormCounts(sim2)
sim2 <- runPCA(sim2)
plotPCA(sim2, colour_by = "Step") + ggtitle("Lots of steps")
3.9.3 path.skew - Path skew
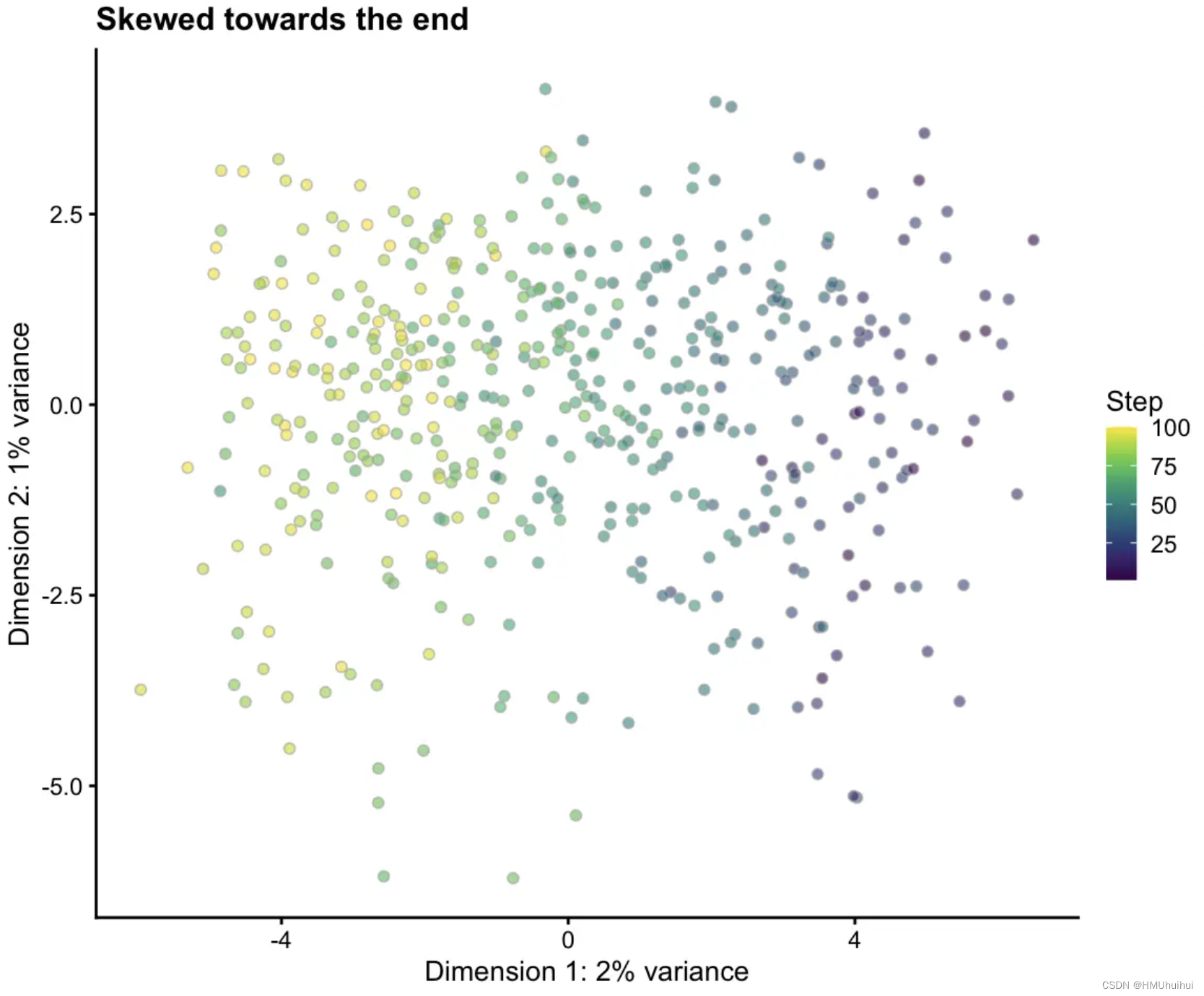
默认情况下,细胞沿一条路径均匀分布,但有时在分布中引入歪斜(skew)是有用的,例如,您可能想要模拟干细胞较少而分化细胞较多的场景。将path.skew设置为0将意味着所有细胞都来自终结点,而更高的值(最高为1)将使它们向起始点倾斜。
# Skew towards the end
sim1 <- splatSimulatePaths(params.groups, path.skew = 0.1,
de.prob = 0.5, de.facLoc = 0.2, verbose = FALSE)
sim1 <- logNormCounts(sim1)
sim1 <- runPCA(sim1)
plotPCA(sim1, colour_by = "Step") + ggtitle("Skewed towards the end")
3.9.4 path.nonlinearProb - Non-linear probability
大多数基因是沿着一条路径以线性方式插入的,但在现实中,情况可能并不总是如此。例如,很容易想象这样一个基因,它在一个过程开始时低表达,在中间高表达,在结束时低表达。path.nonlinearProb参数控制基因沿路径以非线性方式改变的概率。
3.9.5 path.sigmaFac - Path skew
沿着路径的非线性变化是通过在两个端点之间建立布朗桥(Brownian bridge)来实现的。布朗桥是一种布朗运动,其控制方式是端点是固定的。path.sigmaFac参数控制布朗运动中每一步的极端程度,因此插值(interpolation)与线性路径的差别有多大。















 65
65











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









