






宏基因组二代测序技术(metagenomics next-generation sequencing,mNGS)是以特定环境中的整个微生物群落作为研究对象,它摆脱了微生物难分离难培养的技术限制,只需从样品中提取全部微生物的总DNA进行测序。宏基因组二代(医学)是针对医学研究中的样本(粪便、肠道、体液、组织等)进行宏基因组测序,从基因组水平上揭示微生物菌群的物种及基因功能信息,探索其与临床及宿主之间的关系。针对医学样品,美格基因推出宏基因组二代(医学)测序分析服务产品。通过对医学样品的微生物群落结构和功能的解析,研究和理解微生物在人类健康与疾病中的作用。
文章一

英文标题:Consistent signatures in the human gut microbiome of old- and young-onset colorectal cancer
中文标题:老年和青年发病结直肠癌患者肠道微生物组的一致特征
期刊:Nature Communications
发表时间:2024-04-22
样品类型:粪便
组学策略:宏基因组
链接:https://doi.org/10.1038/s41467-024-47523-x
摘要:近几十年来,年轻发病结直肠癌(yCRC)的发病率有所上升,但关于这些患者的肠道微生物组知之甚少。大多数研究都集中在老年发病结直肠癌(oCRC)上,目前尚不清楚从老年患者中得出的CRC特征是否适用于年轻患者。为解决这一问题,坐着从两个独立的队列中收集了迄今为止最大的yCRC肠道宏基因组数据,并发现CRC肠道微生物组与成年期年龄之间的关联有限。差异分析显示,已知与CRC相关的类群,如共生梭菌(Clostridium symbiosum)、口腔链球菌(Peptostreptococcus stomatis)、微小帕夫龙斯菌(Parvimonas micra)和哈特韦氏(Hungatella hathewayi),在老年和年轻发病患者中均显著富集(假发现率<0.05)。观察到核梭杆菌(Fusobacterium nucleatum)、脆弱拟杆菌(Bacteroides fragilis)和大肠埃希菌(Escherichia coli)在oCRC和yCRC中的菌株水平模式相似。几乎所有与oCRC相关的宏基因组途径在年轻患者中也显示出方向一致的变化。重要的是,与CRC相关的毒力因子(如fadA、bft)在oCRC和yCRC中均比各自对照组更丰富。此外,基于微生物组的分类模型对老年和年轻发病患者的CRC状态预测准确性相似,强调了不同年龄组间微生物特征的一致性。
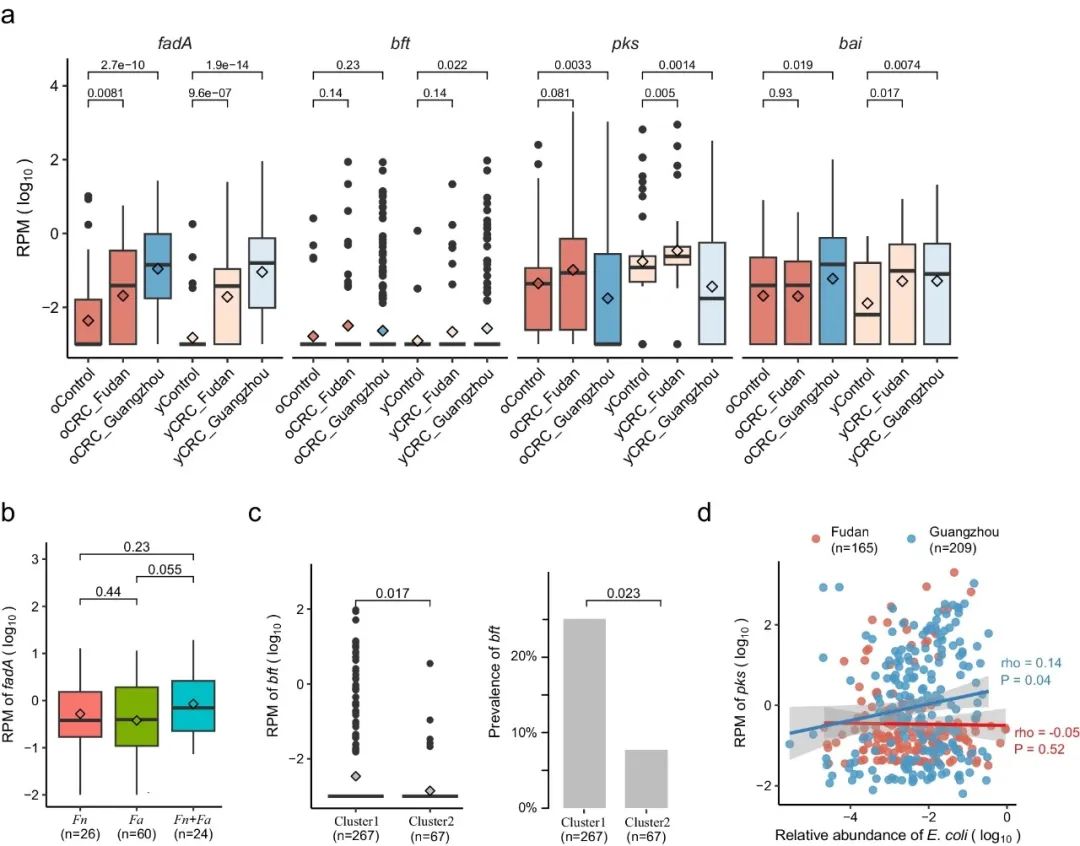
图1:两个独立队列中老年和年轻发病患者CRC相关毒力因子和毒素的富集
文章二

英文标题:Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults
中文标题:针对重症成人群体的脓毒症诊断:结合宿主-微生物血浆宏基因组学的前瞻性队列研究
期刊:Nature Microbiology
发表时间:2022-10-20
样品类型:血液
组学策略:宏基因组、转录组
链接:https://doi.org/10.1038/s41564-022-01237-2
摘要:作者对住院后的重症患者进行了全血(n=221)和血浆(n=138)样本的综合宿主及病原体宏基因组RNA和DNA的下一代测序(mNGS)。根据临床和微生物学标准,作者将患者分为脓毒症组。利用全血基因表达数据,通过训练有素的装袋支持向量机(bSVM)分类器区分了脓毒症患者与非感染性系统性炎症状况患者(训练集下的受试者工作特征曲线面积(AUC)=0.81;验证集下的AUC=0.82)。血浆RNA同样揭示了脓毒症的转录特征,包含多个先前报道的脓毒症生物标志物基因,并建立了一个bSVM脓毒症诊断分类器(训练集AUC=0.97;验证集AUC=0.77)。根据病原体及其感染部位的不同,血浆mNGS的病原体检测性能有所变化。为了提高病毒的检出率,作者开发了二级转录组分类器(训练集AUC=0.94;验证集AUC=0.96)。作者结合宿主和微生物特征,建立了综合脓毒症诊断模型,该模型识别出了99%经微生物学确认的脓毒症病例,同时在疑似脓毒症病例中预测脓毒症的比例为74%,在不确定脓毒症病例中为89%。总之,作者建议将宿主转录组分析与来自核酸的广谱宏基因组病原体检测相结合,是脓毒症诊断的一个有前景的工具。
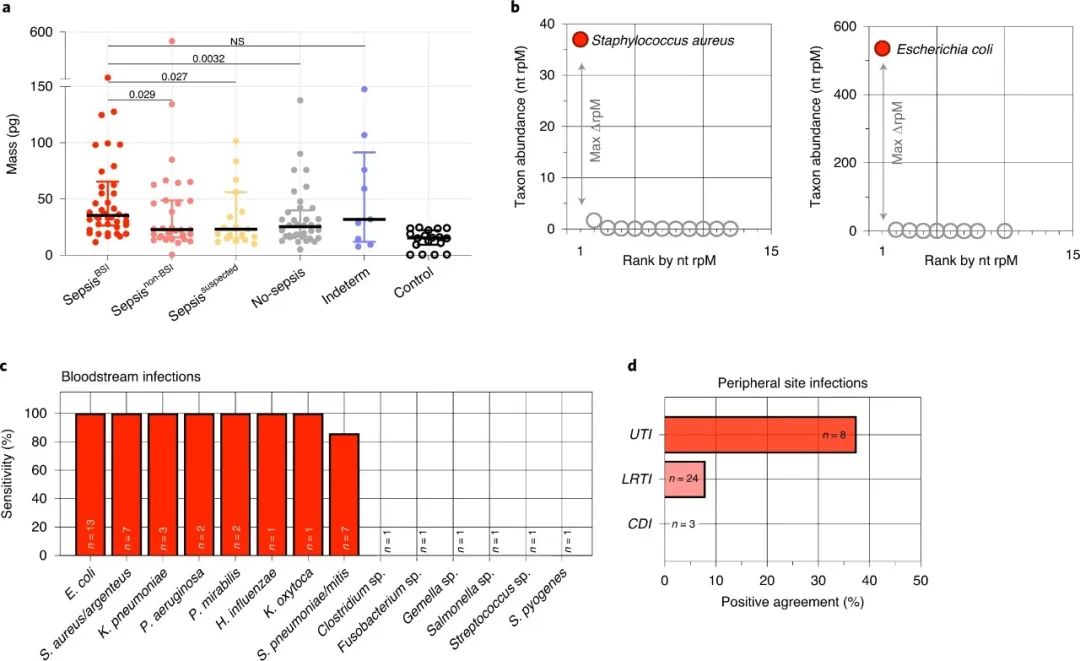
图2:血浆mNGS检测脓毒症病原体
文章三

英文标题:Using 16S rDNA and metagenomic sequencing technology to analyze the fecal microbiome of children with avoidant/restrictive food intake disorder
中文标题:应用16S rDNA和宏基因组测序技术分析回避/限制性食物摄入障碍儿童的粪便微生物组
期刊:Scientific Reports
发表时间:2023-11-22
样品类型:粪便
组学策略:宏基因组、16S
链接:https://doi.org/10.1038/s41598-023-47760-y
摘要:本研究旨在探讨回避/限制性食物摄入障碍(ARFID)儿童的肠道微生物分布及其功能。共招募了135名儿童参与研究,其中包括102名ARFID患儿和33名健康儿童。通过16S rDNA和宏基因组测序分析粪便样本,以探究ARFID组与健康对照(HC)组之间肠道微生物组成、多样性及功能的差异。研究发现,ARFID儿童的肠道微生物组成和多样性与健康儿童存在差异,但3至6岁和7至12岁两个年龄段的ARFID儿童之间,其肠道微生物组成和多样性并无显著不同。在门水平上,通过16S rDNA和宏基因组测序鉴定出的两组中最丰富的微生物相同。而在属水平上,虽然Bacteroides在ARFID组中的丰度较高(P>0.05),但宏基因组测序结果显示,与HC组相比,ARFID组中Bacteroides的丰度显著增加(P=0.041)。在种水平上,ARFID组中最丰富的菌种为大肠埃希氏菌、嗜热链球菌和Lachnospira eligens,而HC组中则以Prevotella copri、双歧杆菌假链状亚种和Ruminococcus gnavus为主;然而,这些菌种在两组间的丰度无统计学显著差异。通过LefSe分析发现,ARFID组中肠杆菌目及相应科Enterobacteriaceae、Bacteroidaceae科及其对应属Bacteroides、以及Bacteroides vulgatus种的丰度较高;相比之下,HC组中显著富集了放线菌门、放线菌纲、双歧杆菌目、双歧杆菌科以及双歧杆菌属。Y1和Y2两组间(按年龄分)的Chao1、Shannon和Simpson指数无显著差异。在门水平上,两组中均以厚壁菌门、拟杆菌门、变形菌门和放线菌门为最丰富类群,但各菌群丰度无显著差异。属水平上,Y2组中的Faecalibacterium较Y1组更为丰富,且差异具有统计学意义(P=0.037)。基于KEGG注释,ARFID儿童与健康儿童的肠道微生物功能无显著差异;但从CAZy数据库分析发现,GT26在ARFID儿童中显著富集。在抗生素抗性基因方面,ARFID组中最丰富的基因包括vanT、tetQ、adeF和ermF,且该组中大环内酯类抗生素抗性基因的丰度显著高于HC组(P=0.041)。
综上所述,与健康儿童相比,ARFID儿童的肠道微生物分布及功能基因存在差异,提示肠道微生物组可能在ARFID的发病机制中扮演重要角色。
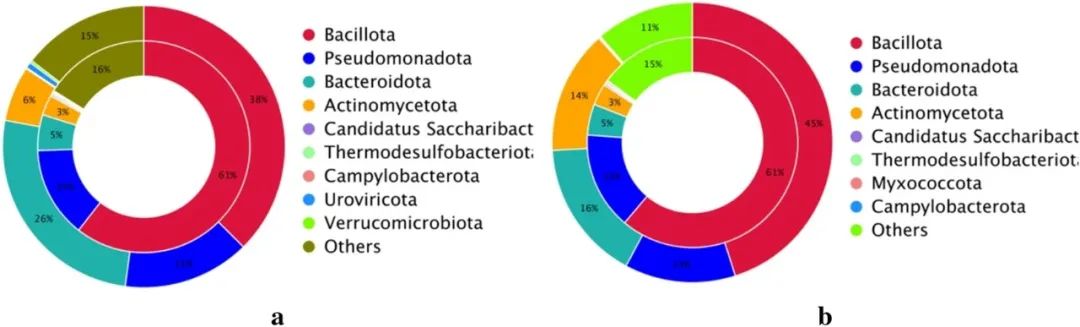
图3:两组耐药基因与物种归因关系的双圆图
文章四

英文标题:Comparative characterization of the infant gut microbiome and their maternal lineage by a multi-omics approach
中文标题:通过多组学方法对婴儿肠道微生物组及其母系进行比较表征
期刊:Nature Communications
发表时间:2024-04-08
样品类型:粪便
组学策略:宏基因组、代谢组、16S
链接:https://doi.org/10.1038/s41467-024-47182-y
摘要:人类肠道微生物群在婴儿期建立并成熟,这一阶段的失调可能会导致以后的生活中出现病理。我们进行了一项包括三代家庭成员的多组学研究,以调查肠道微生物群的早期发展。来自200个个体的粪便样本,包括婴儿(0-12个月大;55%女性,45%男性)及其各自的母亲和祖母,使用两个独立的代谢组学平台和宏基因组学进行了分析。对于代谢组学,应用了气相色谱和毛细管电泳耦合质谱。对于宏基因组学,同时进行了16S rRNA基因和猎枪测序。在这里,我们显示婴儿在粪便微生物群、功能和代谢组方面与他们的长辈有很大差异。婴儿的微生物群不如成年人多样化,并且在几个代谢物类别中存在差异,例如短链和支链脂肪酸,这与细菌种群的变化有关。这些发现为同一世代内肠道微生物群的形成提供了创新的生化见解,这可能有助于改善儿童健康结果。
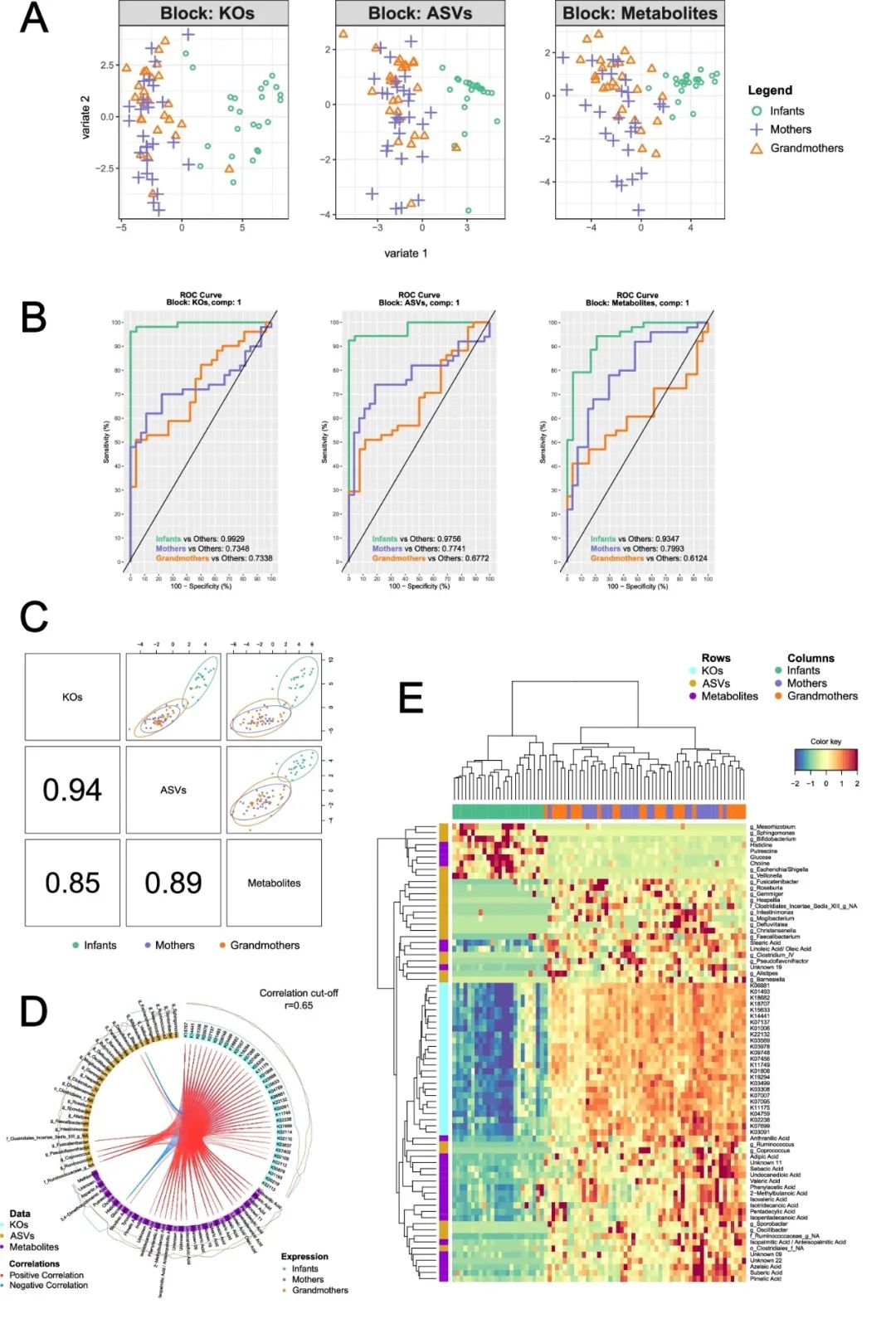
图4:使用mixOmics和DIABLO集成三个组学
文章五

英文标题:Gut Bacteroides act in a microbial consortium to cause susceptibility to severe malaria
中文标题:肠道类杆菌在微生物群中起作用,导致对严重疟疾的易感性
期刊:Nature Communications
发表时间:2023-10-13
样品类型:盲肠、血液
组学策略:宏基因组、转录组、16S
链接:https://doi.org/10.1038/s41467-023-42235-0
摘要:疟疾是由疟原虫引起的,仍然是全球发病率和死亡率的重要原因。肠道细菌可以影响疟疾的严重程度,但特定细菌对严重疟疾风险的贡献尚不清楚。在这里,多组学方法表明特定种类的拟杆菌与严重疟疾的风险有因果关系。用小鼠分离的脆弱拟杆菌灌胃的约氏疟原虫hyperparasitemia-resistant小鼠会发生约氏拟杆菌高寄生虫血症。此外,在患有严重疟疾贫血的乌干达儿童中,拟杆菌明显多于无症状的恶性疟原虫感染。人类分离的卡氏拟杆菌、均匀拟杆菌和卵形拟杆菌能够导致小鼠对严重疟疾的易感性。虽然单独使用拟杆菌的无菌小鼠的单核化不足以导致对高寄生虫血症的易感性,但多项研究的荟萃分析支持拟杆菌在严重疟疾易感性中的主要作用。针对肠道拟杆菌的方法为预防严重疟疾和相关死亡提供了机会。
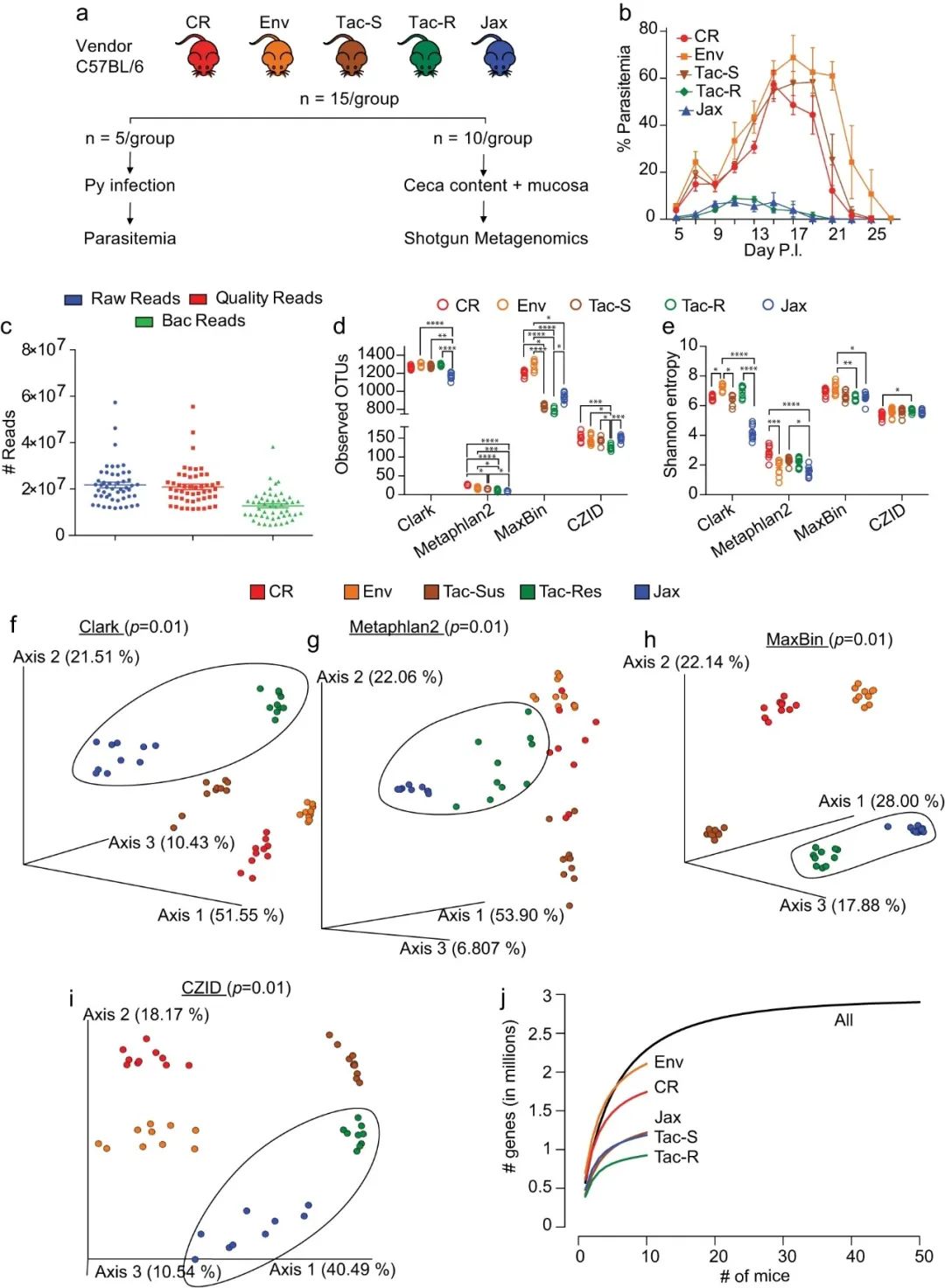
图5:鸟枪宏基因组学揭示了高寄生虫血症耐药和易感小鼠体内和之间不同的肠道微生物群组成和遗传潜力
参考文献
[1] Qin, Y., Tong, X., Mei, WJ. et al. Consistent signatures in the human gut microbiome of old- and young-onset colorectal cancer. Nat Commun 15, 3396 (2024).
[2] Kalantar, K.L., Neyton, L., Abdelghany, M. et al. Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults. Nat Microbiol 7, 1805–1816 (2022).
[3] Ye, Q., Sun, S., Deng, J. et al. Using 16S rDNA and metagenomic sequencing technology to analyze the fecal microbiome of children with avoidant/restrictive food intake disorder. Sci Rep 13, 20253 (2023).
[4] Barker-Tejeda, T.C., Zubeldia-Varela, E., Macías-Camero, A. et al. Comparative characterization of the infant gut microbiome and their maternal lineage by a multi-omics approach. Nat Commun 15, 3004 (2024).
[5] Mandal, R.K., Mandal, A., Denny, J.E. et al. Gut Bacteroides act in a microbial consortium to cause susceptibility to severe malaria. Nat Commun 14, 6465 (2023).















 712
712

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









