






物种/功能丰度表是宏基因组分析结果中最核心的文件,表格第一行是样品名称,第一列是物种或功能描述,其余部分是物种/功能在对应样品中的相对丰度,如下图(数据来自美格基因云平台的宏基因组demo项目)所示。
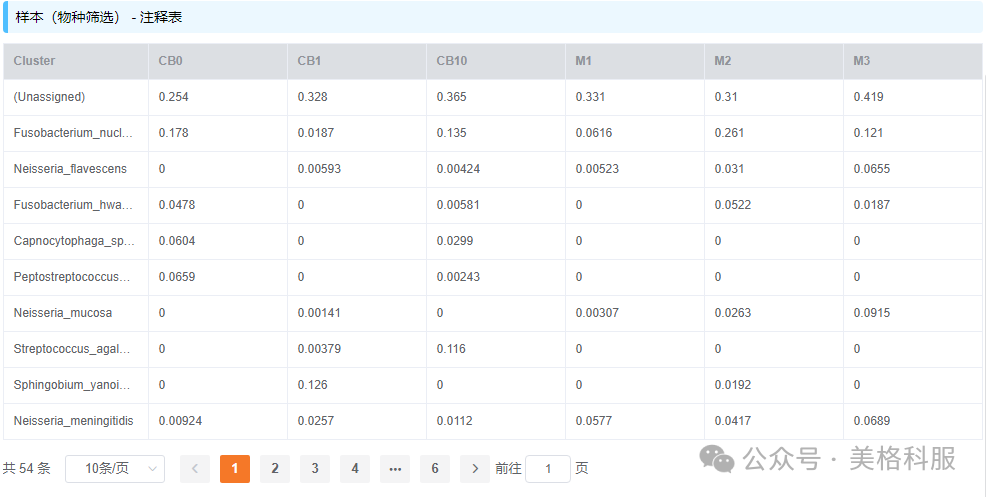
图1 物种(种水平)丰度表
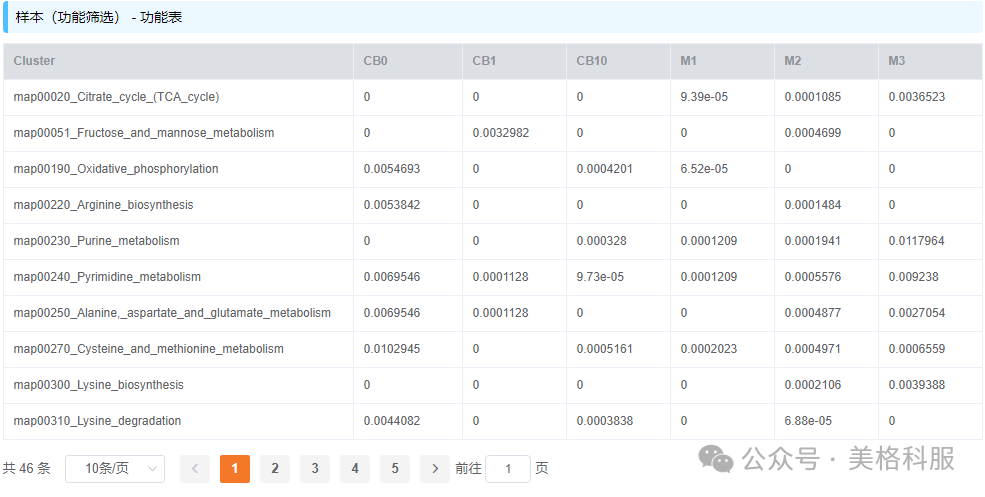
图2 功能(KEGG通路)丰度表
针对这个丰度表
不同的数据可视化方法
可以用来解释不同的问题
1.样本中丰度最高的物种/功能是哪几个?
答:可以通过将丰度排序找出丰度最高的物种/功能,然后使用柱状图来展示结果。
可视化R代码如下:

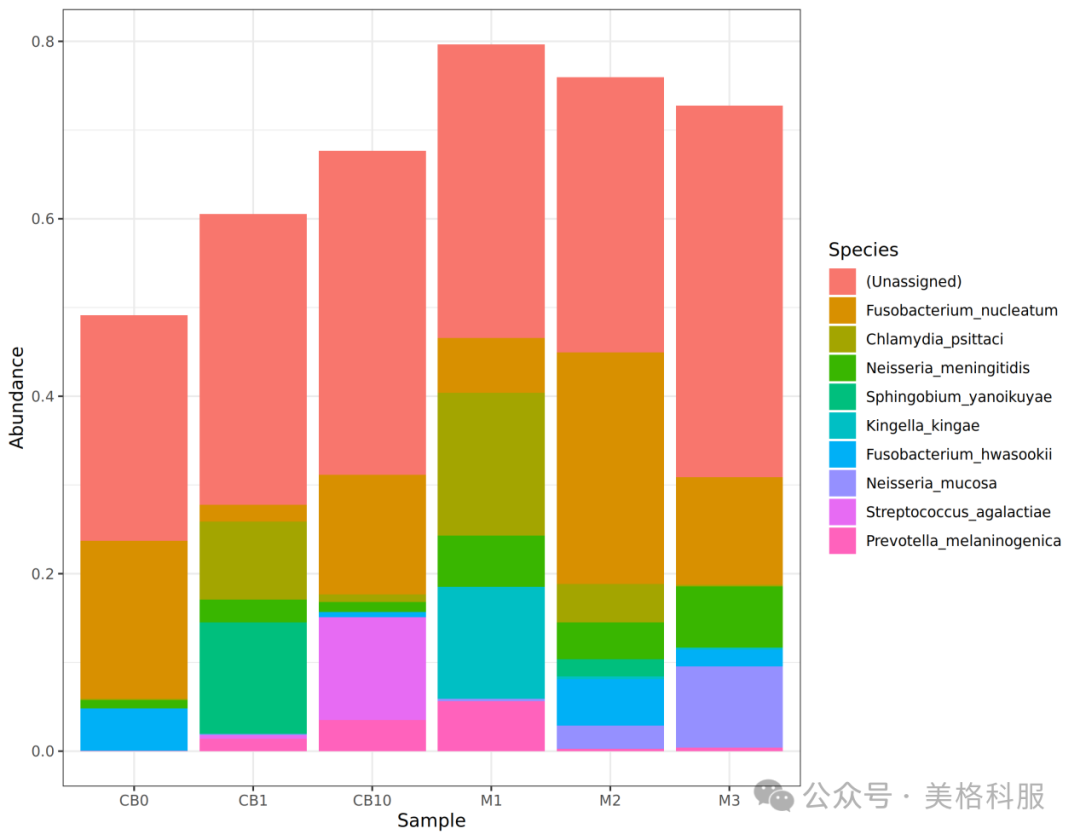
图3 物种丰度前top10柱状图
2.在物种/功能的数量上,
哪个样本多?哪个样本少?
不同样本间共有的物种/功能数量是多少?
答:首先,判断一个样品中是否有某个物种/功能的依据是丰度不为0。所以要回答这些问题,需要分别统计表格里的每个样品(每一列)丰度不为0的物种/功能数量,以及统计不同样品间物种/功能的交集数量。数据统计和可视化的都可以由R包VennDiagram来完成。
可视化R代码如下:

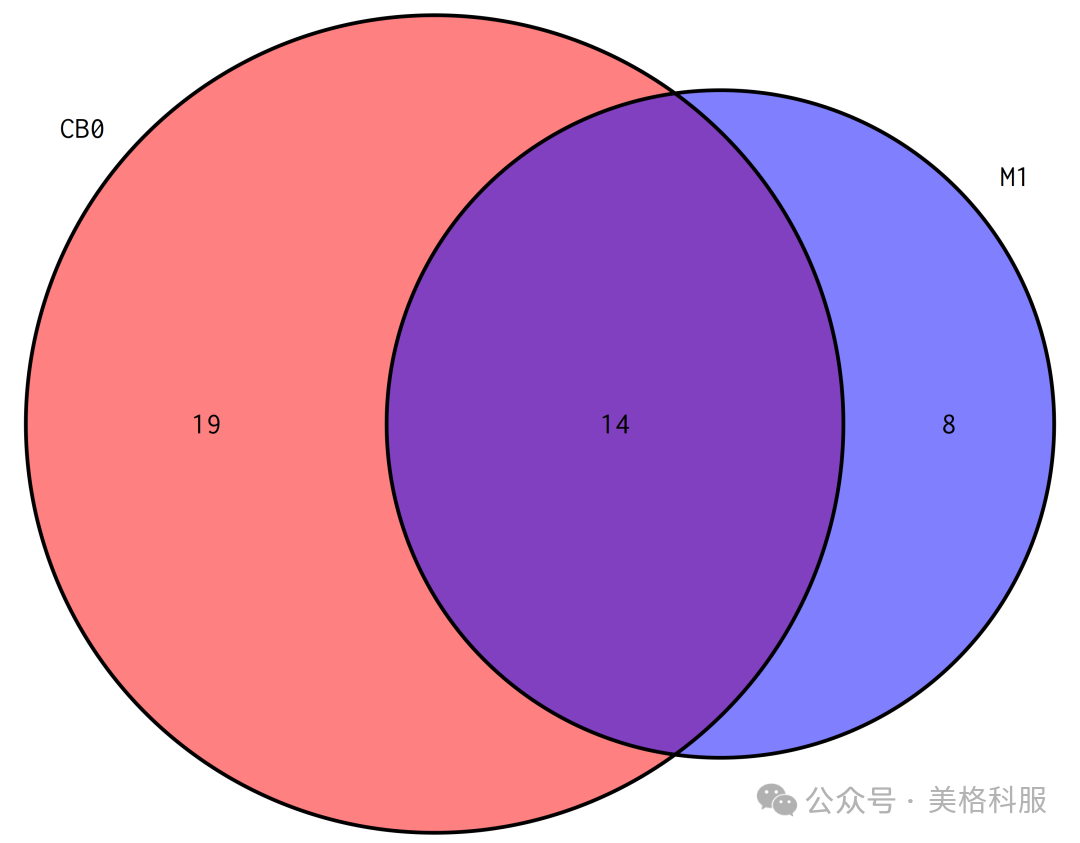
图4 物种数量Venn图
3.在丰度组成上,哪些样本比较相似?
哪些物种/功能比较相似?如何更直观的了解相同物种/功能在不同样本中的丰度差别?
答:适合用来回答这些问题的可视化方法为聚类热图。聚类热图中的颜色深浅反映了标准化后的丰度大小,样本聚类树反映了样本间距离,物种/功能聚类树反映了物种/功能间的距离,靠的近的丰度组成越相似。数据标准化、样本间距离计算与聚类、物种/功能间距离计算与聚类,都可以由R包pheatmap来完成。
可视化R代码如下:

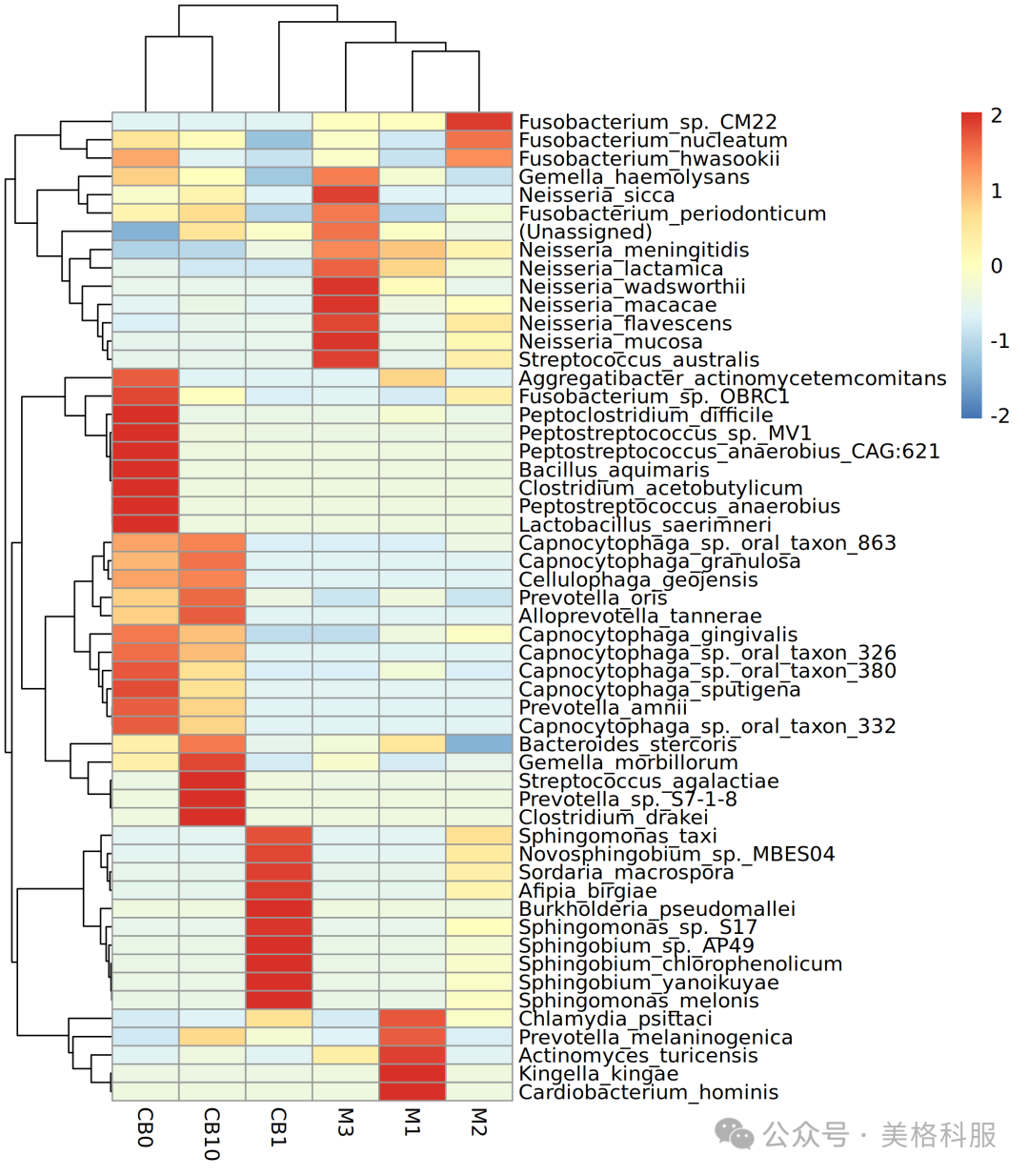
图5 物种丰度聚类热图


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









