






持续更新中ing,如有不正确的地方,谢谢指正!
CRAN地址:https://cran.r-project.org/web/packages/vcfR/readme/README.html
GITHUB地址:https://knausb.github.io/vcfR_documentation/
参考文献:“VCFR: a package to manipulate and visualize variant call format data in R”
GB/T 7714引用格式:Knaus B J, Grünwald N J. vcfr: a package to manipulate and visualize variant call format data in R[J]. Molecular ecology resources, 2017, 17(1): 44-53.
文章目录
一、预备知识
VCF格式数据是什么
VCF(variant call format):VCF文件可作为文本文件或压缩文件提供,典型的格式:gzip,常用于生物信息学和遗传分析。已成为对变异数据进行下游分析的标准,分析VCF数据之前,应根据SAMTOOLS或GATK-HC等变异调用提供的质量指标对遗传变异进行筛选。
VCF格式数据都以一个meta区域开始:定义了文件数据部分中的缩写,每行提供一个定义。还包含一个表格部分:每一行代表一个变异,前八列包含描述每个变异的信息,第九列为基因型记录的FORMAT规范,剩余列为样本信息(每一列对应一个样本,其值由基因型和FORMAT列指定的信息组成)。VCF格式不是严格的表格数据形式。
不同的变异可能包含不同的FORMAT数据,这种情况便要对变异进行独立处理。FORMAT允许每个基因型包含灵活的数量信息。
变异数据包括:SNP、缺失、插入、大结构变异、复杂事件等。
VCF文件详细格式介绍:ADNI VCF数据介绍
R语言知识
R语言是一种处理遗传数据的流行环境。
二、vcfR包
主要介绍一下vcfR包功能!
vcfR提供了一种可以快速开发质量指标的过滤器的工具,可以轻松地针对单个项目或实验设计进行定制。为了减少计算时间,vcfR的关键组件是在C++中实现的,从R中调用。
理想情况,VCF文件处理 :可快速的输入和输出、能读取文本和压缩文件、处理非表格VCF规范、可选文件子集进行输入(对感兴趣的行和列进行子集扫描)。在数据集上进行操作。可能需要比给定的可用计算环境更多的物理内存。
通常使用一组质量控制标准来过滤数据。有图形和交互式的方法可以快速确定过滤中的选择如何影响结果数据集。
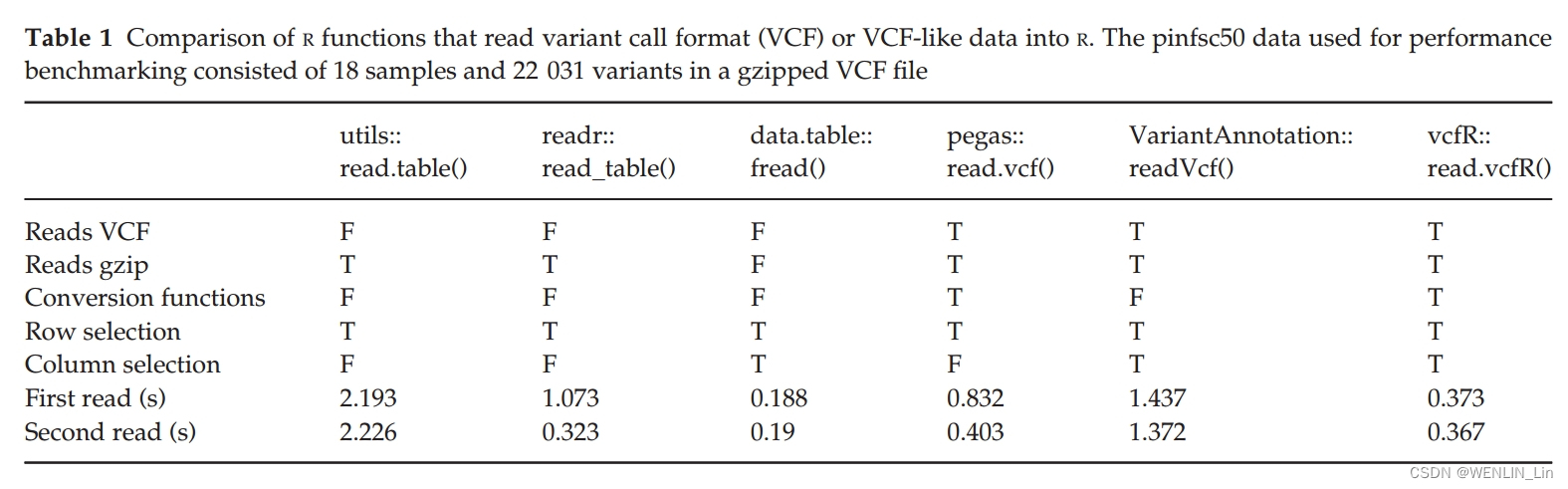
处理VCF数据不同的包进行对比
在R中,虽然已经有很多处理VCF格式数据的包,但仍存在一些不足。以下给出一些vcfR与其他包进行比较的优缺点。
| vcfR包 | 读取速度与data. Table::fread()和readr::read_table() 相当;是否压缩都可以读取;信息保持完整,可根据需要进行解析 ,灵活提取行和列,节省内存;转换函数支持转换为其他R遗传包支持的格式;输入包括质量指标用于后续过滤 | |
|---|---|---|
| utils::read.table() | 可跳过meta部分进行读取 | 性能不佳,读取速度慢 |
| readr::read_table() | 更有效率一些比上一个方法 | – |
| data.table::fread() | 可跳过meta部分进行读取 ,可灵活访问行和列,性能最好,读取速度快,比第一种效率高 | 不能对压缩包gzipped进行读取,只能读取表格数据。在unix系统中可以通过修改调用zcat或gzcat来读入压缩数据,window用户只能解压文件 |
| PopGenome::readVCF() | 经过faidx索引和bgzip压缩读入VCF文件,未来高吞吐量项目可能采取的方向(目前gzip文件最常见) | 不支持将数据转为其他R的格式 |
| Bioconductor包的VariantAnnotation::readVcf() | 解析INFO列中的数据,并将gt部分数据解析为单独的矩阵,比其他选项执行更多的工作来提供“完整服务” | 与utils::read.table()读取速度接近,似乎不如其他选项→生成数据结构较复杂,数据结构比其他选项更完整,并且似乎是Bioconductor项目特有的 |
| pegas::read.vcf() | 满足所有标准 | 只读取基因型,不读取与基因型相关的数据,不会为质量过滤提供信息 |
| VCFTOOLs | 提供了一套广泛的数据过滤和分析工具 ,是命令行工具 | 适合缺乏图形用户界面或缺乏交互式可视化的基于云的排队系统中实现的高性能计算环境 |
vcfR包的优点:
- 交互式,可操作和可视化VCF数据的R包,
- 高效读写,有效的读取VCF文件到内存并写回磁盘,并可转换为R中其他遗传学使用的数据格式
- 解析功能,有效的提取基因型矩阵或其他相关信息
- 绘图函数,快速、直观地评估变量特征
- 对R中提供的大量统计和图形工具的随时访问
- 质量过滤
- 保持数据的完整性,并可灵活提取信息。允许用户只提取并解析感兴趣的信息,提供灵活性。
其他包:
- 有些只能提取基因型,不能提取描述基因型质量的相关数据,意味着不能提供质量过滤的相关信息,而质量过滤是获得高质量数据的关键任务。
通过输入两次数据确定函数是否实现了某种形式的缓存,这可能会提高第二次读取的时间。时间用microbenchmark::microbenchmark()或system.time()计算
较大的数据集可以使用read.vcfR()函数中的列和行选择作为子集。理论上任何产生VCF文件的基因组或亚基因组项目都可以用VCFR进行处理。
三、vcfR包含的函数
由于数据不是严格的表格格式,因此需要进一步处理才能访问数据。
extract.gt()
extract.gt():从VCF数据的gt部分解析元素。快速提取与指定的FORMAT值之一对应的字符串或数字矩阵,然后对这些矩阵进行操作,并可视化。
extract.gt(
x,
element = "GT",
mask = FALSE,
as.numeric = FALSE,
return.alleles = FALSE,
IDtoRowNames = TRUE,
extract = TRUE,
convertNA = TRUE
)
| x | vcfR或chromR数据 |
|---|---|
| element | 从vcf基因型数据中提取元素,常用选项:“DP”、“GT”和“GQ” |
| mask | 逻辑值,是否应用掩码(TRUE)或返回所有变量(FALSE)。或者可以提供逻辑向量。 |
| as.numeric | 逻辑值,矩阵转为数字 |
| return.alleles | 逻辑值,是否返回基因型(0/1)或等位基因(A/T) |
| IDtoRowNames | 逻辑值,是否使用fix区域中的ID列作为行名 |
| extract | 逻辑值,返回提取的元素还是返回剩余字符串的逻辑 |
| convertNA | 逻辑值,是否转换‘.’为NA |
| unphased_as_NA | 如何处理未分阶段的基因型 |
| verbose | 是否生成详细输出 |
| return.indels | 逻辑值,是否返回indels |
mask参数允许在使用chromR对象时实现mask。
“as.numeric”选项将结果从字符转换为数字。请注意,如果数据实际上不是数字,它将导致可能无法解释的数字结果。
“return.alleles”选项允许将数字编码的基因型(例如,0/1)的默认行为转换为它们的核酸表示(例如,A/T)。请注意,这不是用于正则表达式,因为在其他函数中使用了类似的参数。
Extract允许用户仅提取指定的元素(TRUE)或除指定元素之外的每个元素。
请注意,当’as.numeric’设置为’TRUE’,但数据实际上不是数值,意外的结果可能会发生。例如,基因型字段通常会填充诸如“0/1”或“0”之类的值。虽然它们可能显示为数字,但它们包含非数字的分隔符(正斜杠或管道)。这意味着不存在直接转换为数字的情况,应该预期会出现意外值。
chromoqc()
chromoqc()可用于可视化chromR对象。
后续的汇总统计(杂合度,有效大小),滑动窗口分析(核苷酸含量,注释位置数量,每个窗口的变异),可以以表格格式储存,并使用graphics::hist()或graphics::plot())进行可视化,或者保存到文件中。
masker()
masker():可以对DP、MQ等的阈值进行过滤
write.vcf()
导出vcf数据,在VCF文件上操作的任何软件可以用于下游分析、
vcfR2genind()
可用于将适量的VCF数据转换为genind对象,从而允许在adegenet中进行分析
as.genclone()
使用poppr::as.genclone()可以很容易地将Genind对象转换为genclone对象,以便在poppr中进行分析
vcfR2genlight()
ADEGENET包创建了专门用于高通量测序应用的genlight对象。使用vcfR2genlight()函数从VCFR类的对象创建genlight类的对象。
poppr::as.snpclone()
一旦创建了genlight类的对象,就可以使poppr::as.snpclone()将其转换为snpclone类的对象。
vcfR2DNAbin()
当提供序列信息后,可以使用vcfR2DNAbin()将VCF数据转换为DNAbin类对象,以便在APE或PEGAS中进行分析。
四、示例数据集(pinfsc50)
pinfsc50为二倍体卵菌致病菌疫霉数据,用来说明vcfR的应用。包含丰富的描述信息如:质量指标等的文件可提供有指导意义的示例。
当包超过5MB时,在CRAN上会产生“NOTE”。所以pinfsc50数据集以包的形式发布。
数据集包含29行的meta信息,22031个变异行和27列(18个样本)
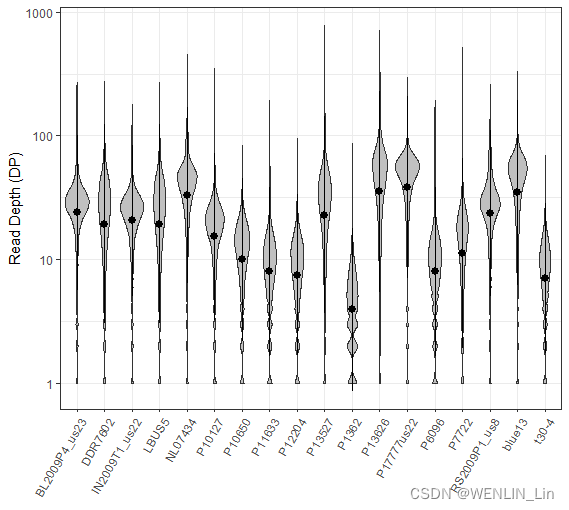
示例:提取变异中的DP(读取深度)得到一个数值矩阵,列为样本,行为变异,再将这些数据可视化(使用ggplot2)。
library(vcfR)
library(ggplot2)
library(pinfsc50)
library(reshape2)
vcf <- system.file('extdata','pinf_sc50.vcf.gz',package = 'pinfsc50')
vcf <- vcfR::read.vcfR(vcf, verbose = FALSE)
dp <- extract.gt(vcf, element='DP', as.numeric = TRUE)
#数据重组
dpf <- melt(dp, varnames=c('Index', 'Sample'),
value.name = 'Depth', na.rm=TRUE)
dpf <- dpf[ dpf$Depth > 0,]
p <- ggplot(dpf, aes(x=Sample, y=Depth))
+ geom_violin(fill='#C0C0C0', adjust=1.0,scale = 'count', trim=TRUE)
+ theme_bw()
+ ylab('Read Depth (DP)')
+ theme(axis.title.x = element_blank(),axis.text.x = element_text(angle = 60, hjust = 1))
+ stat_summary(fun.data=mean_sdl,geom='pointrange', color='black')
+ scale_y_continuous(trans=scales::log2_trans(),breaks=c(1, 10, 100, 1000))

















 3832
3832

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









