







如果您在生物信息分析过程中,遇到计算资源不足环境依赖时常报错,找不到学习社群。那么快来试试我们的,生信专用云服务器祝您
一.什么是ATAC-Seq
ATAC-Seq 是“Assay for Transposase-Accessible Chromatin with high-throughput Sequencing”的缩写。 ATAC-Seq 方法依赖于使用高活性转座酶 Tn5 的下一代测序(NGS)文库的构建。将 NGS 接头连接到转座酶上,该转座酶可以使染色质断裂并同时将这些接头整合到开放的染色质区域中。构建的文库可通过 NGS 测序,并使用生物信息学分析具有可及或可访问染色质的基因组区域。
尽管 ATAC-seq 实验方法相对简单且稳定,但是专门为 ATAC-seq 测序数据开发的生物信息学分析软件却非常少。由于 ATAC-seq 和 ChIP-seq 数据的相似性较高,ChIP-seq 分析使用的软件一般也可用于 ATAC-seq 的分析,但是使用 ChIP-seq 软件分析得到的 ATAC-seq 结果尚未得到系统性的评估。
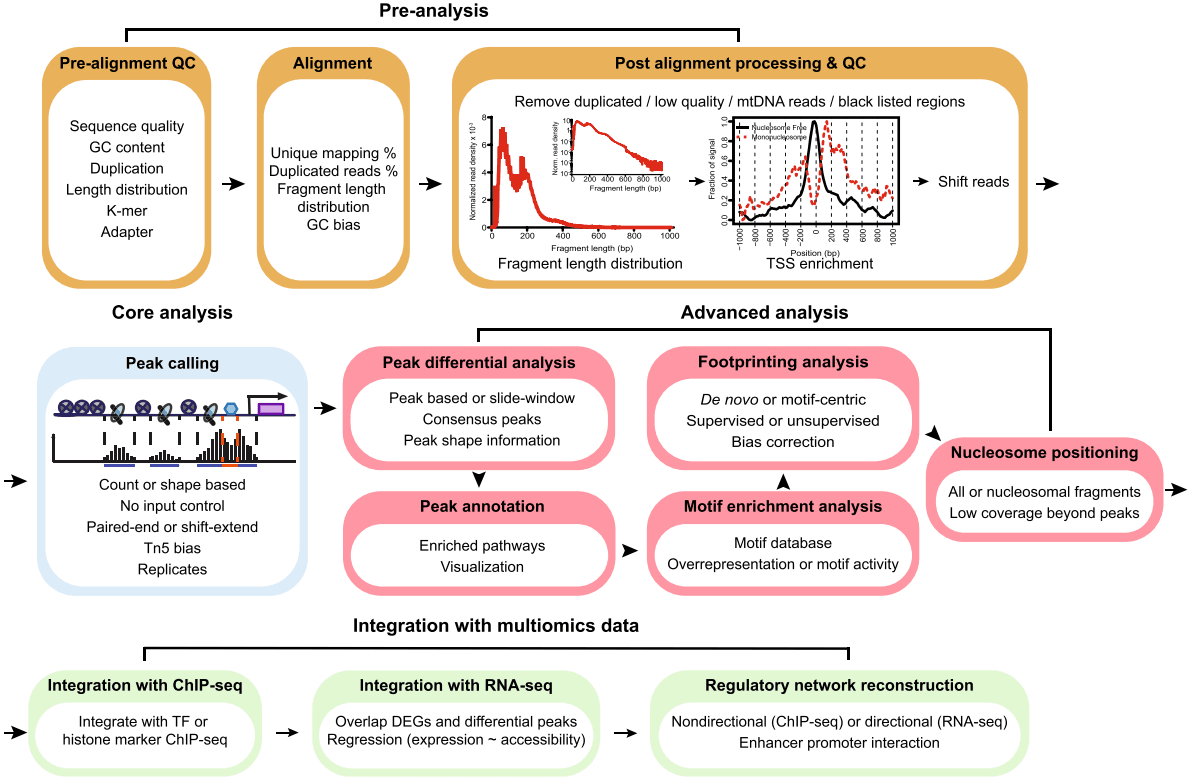
- 质控:使用 FastQC 对测序数据进行质量检测。
- 比对:利用 BWA 将测序数据比对到参考基因组。
- 峰值召回:使用 MACS2 进行 Peak Calling。
- 峰值注释:使用 ChIPseeker 对 peak 位置及邻近基因进行注释。
- 差异分析:分析不同样本或条件下的 peak 差异。
- 基序分析:识别并分析基因组区段中的基序。
- 多组学分析整合:将 ATAC-Seq 数据与其他组学数据(如 RNA-seq)进行整合分析。
二.版本信息
- 成都理工大学超算平台 Red Hat 4.8.5-36
- conda 4.10.3
- R 4.4.0
- 测序数据来自 NCBI:PRJNA1080287
- 参考基因组和注释文件来自 Ensembl Plants
- 物种:Oryza sativa Japonica
三.关于一些名称解释
- peaks(峰):在染色质研究中,它用于表示染色质的开放程度。由于测序所得的 reads(读段)集中落在染色质的开放区域,大量堆叠后以可视化的形式呈现出的丰度体现。例如,在某个特定的基因组区域,如果出现明显的 peak,就意味着该区域的染色质开放性较高,可能与基因的活跃表达相关。
- THSs(转座酶超敏感位点):这是对转座酶具有超高敏感性的特定染色质位点。转座酶在此处能够更轻易地发挥作用,导致染色质结构的改变。
- promoter(启动因子):位于结构基因 5’端上游的 DNA 序列,对 RNA 聚合酶起到活化作用,确保其与模板 DNA 准确结合,并赋予转录起始过程特异性。比如在基因表达调控中,不同的启动因子决定了基因转录的起始时机和效率。
- CREs(顺式调控元件):是 DNA 分子中具备转录调节功能的特异序列。在真核基因中,按照功能特性可分为启动子、增强子及沉默子。例如,增强子能够增强基因的转录活性,即使距离基因较远也能发挥作用。
- ACRs(染色质开放区域):指正常或核小体被酶切后,裸露出来的 DNA 片段所在的区域。这些区域通常与基因的表达调控密切相关。
- transposon(转座子):是一段能够从原位置单独复制或断裂下来,环化后插入到另一个位点,并对后续基因起调控作用的 DNA 序列。比如某些细菌中的转座子,能够在基因组中移动,改变基因的排列和功能。
- proximal promoters(近端启动因子):是 DNA 上靠近基因起始位置的一个区域,在此区域蛋白质和其他分子结合,为读取该基因做好准备。
- enhancer(增强子):是远离转录起始点的 DNA 序列,能够决定基因在时间和空间上的特异性表达,增强启动子的转录活性。比如在某些发育过程中,特定的增强子能够激活特定基因在特定组织或细胞中的表达。
- TSS(转录起始位点):在基因内部,其排列顺序通常为转录起始位点(一个碱基)-起始密码子编码序列-终止密码子编码序列-转录终止位点。TSS 是基因转录的起始位置。
- TFs(转录因子):是保证目的基因按特定强度、在特定时间和空间进行表达的蛋白质分子。它们与 RNA 聚合酶Ⅱ共同形成转录起始复合体,参与转录起始过程。
- histone(组蛋白):通常包含 H1、H2A、H2B、H3、H4 等 5 种成分。其中 H1 与 H3 富含赖氨酸,H1 不保守,其他组蛋白的基因则非常保守。除 H1 外,其他 4 种组蛋白以二聚体形式结合形成核小体核心。
- nucleosome(核小体):由 DNA 和组蛋白构成的染色质基本结构单位。每个核小体由 146bp 的 DNA 缠绕组蛋白八聚体 1.75 圈形成,核心颗粒之间通过 50bp 左右的连接 DNA 相连,核小体表面的 DNA 能被特定核酸酶接近并切割。比如在染色体的压缩和基因表达调控中,核小体的结构和分布起着关键作用。
四.软件安装
只需要使用 conda 就可以安装所有需要的软件,主要使用的软件有以下一些:
- sra-tools:快速下载 NCBI SRA 数据
- fastQc:测序数据质量检测与控制
- multiqc:合并质量检测报告
- trim_galore:过滤低质量序列
- bwa:测序数据的回帖
- samtools:对 hisat2 比对的结果进行排序和压缩
- macs2:进行 Peak Calling
- deeptools:计算基因组区段 reads 覆盖度
先创建 conda 虚拟环境,安装所需要的软件,可以自行手动安装,也可以直接导入我的 conda 环境:
git clone https://github.com/yuanj82/NGS-analysis.git
cd NGS-analysis/environment
cp .condarc ~/
conda env create --file atac-seq-env.yml # ATAC-seq创建完成后激活环境就可以使用了:
conda activate atac-seq五.数据获取与预处理
从 NCBI SRA 数据库下载 SRR_Acc_List.txt 文件:
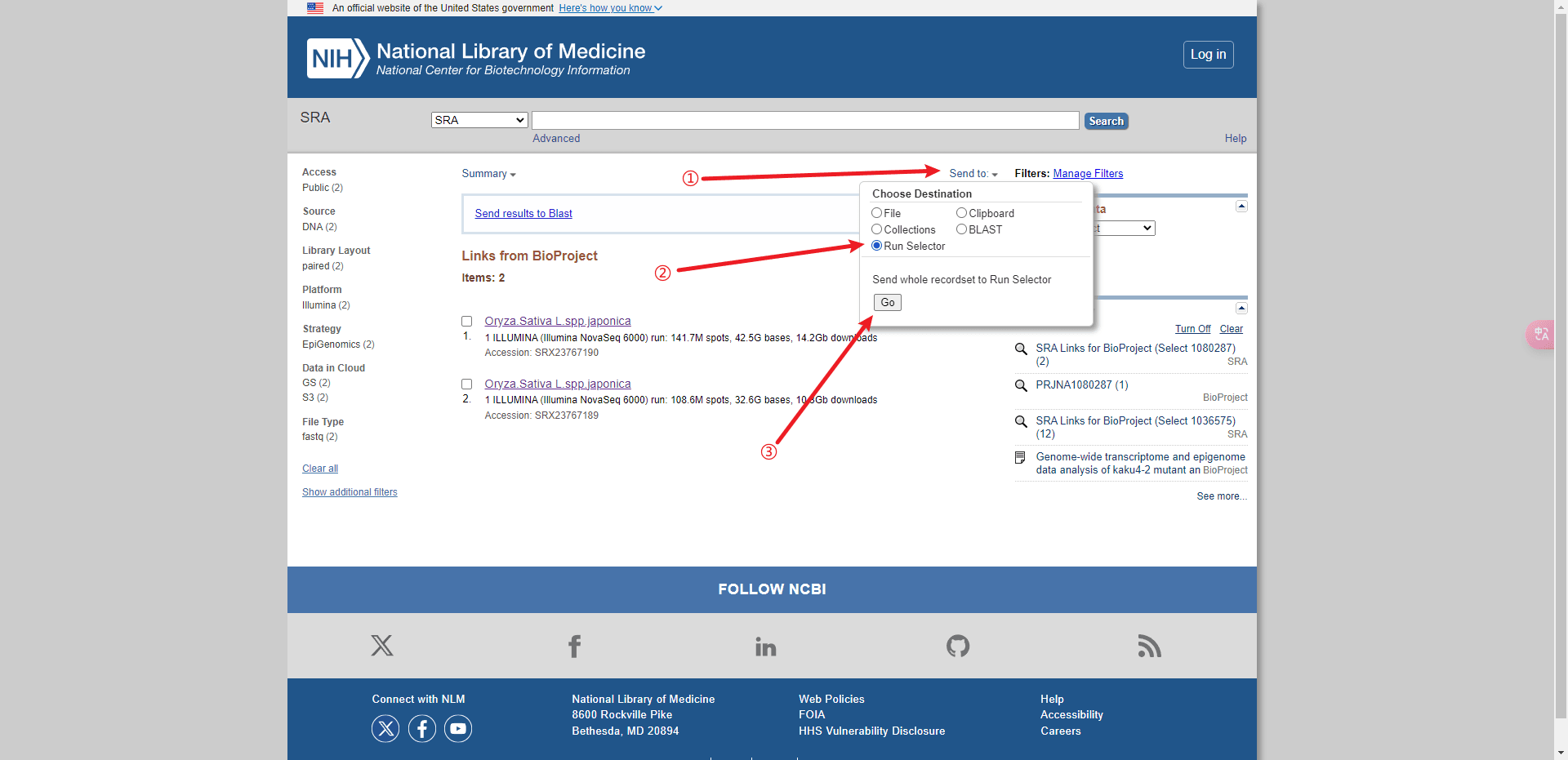
后面的分析都可以基于这个文件进行批处理。
使用 sratools 进行批量下载,下载比较慢,就用 nohup 挂在后台下载,自己可以做点别的事:
nohup prefetch -O . $(<SRR_Acc_List.txt) &下载后的数据是放在一个个 SRRXXXXX 的文件夹里面的,我们用一个小脚本把它全部移动到一个 sra 文件夹便于处理:
mkdir sra
cat ./SRR_Acc_List.txt | while read id; do
mv -f "${id}/${id}.sra" ./sra/
rm -rf "${id}"
done刚刚下载好的数据是 sra 格式的,使用 sratools 将其拆分并输出到 fastqgz 文件夹:
mkdir fastqgz
nohup fastq-dump --gzip --split-3 ./sra/*.sra --outdir ./fastqgz &- –gzip 是将拆分的 fastq 文件压缩归档为 gz 格式
- –split-3 是将文件拆分为正向序列和逆向序列
六.参考基因件以及解释文件
植物的我一般在 Ensembl Plants 下载,用 wget 或 curl 都可以,内存不大,这组数据是水稻的,所以就从 Ensembl Plants 下载水稻的基因组和注释文件:
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/fasta/oryza_sativa/dna/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa.gz
wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-57/gff3/oryza_sativa/Oryza_sativa.IRGSP-1.0.57.gff3.gz解压
gzip -d Oryza_sativa.IRGSP-1.0.57.gff3.gz
gzip -d Oryza_sativa.IRGSP-1.0.dna.toplevel.fa.gz七.使用 FastQc 对测序数据进行质量检测
# 常用参数
fastqc [-o output dir] [–(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
● -o –outdir:FastQC 生成的报告文件的储存路径,生成的报告的文件名是根据输入来定的
● –extract:生成的报告默认会打包成 1 个压缩文件,使用这个参数是让程序不打包
● -t –threads:选择程序运行的线程数,每个线程会占用 250MB 内存,越多越快咯
● -c –contaminants:污染物选项,输入的是一个文件,格式是 Name [Tab] Sequence,里面是可能的污染序列,如果有这个选项,FastQC 会在计算时候评估污染的情况,并在统计的时候进行分析,一般用不到
● -a –adapters:也是输入一个文件,文件的格式 Name [Tab] Sequence,储存的是测序的 adpater 序列信息,如果不输入,目前版本的 FastQC 就按照通用引物来评估序列时候有 adapter 的残留
● -q –quiet:安静运行模式,一般不选这个选项的时候,程序会实时报告运行的状况八.进行质量检测
直接进行批处理:
fastqc ./fastqgz/*.fastq.gz -o ./fastqc_report当然,数据比较多的时候还是挂在后台批处理然后等着就行:
nohup fastqc SRR*.fastq.gz &
multiqc ./fastqc_report # 将多个质量检测报告合并九.使用 Trim Galore 对测序数据进行过滤
trim_galore [options] <filename(s)>
● –quality:设定 Phred quality score 阈值,默认为 20
● –phred33:选择-phred33 或者-phred64,表示测序平台使用的 Phred quality score
● –adapter:输入 adapter 序列。也可以不输入,Trim Galore! 会自动寻找可能性最高的平台对应的 adapter。自动搜选的平台三个,也直接显式输入这三种平台,即–illumina、–nextera 和–small_rna
● –stringency:设定可以忍受的前后 adapter 重叠的碱基数,默认为 1(非常苛刻),可以适度放宽,因为后一个 adapter 几乎不可能被测序仪读到
● –length:设定输出 reads 长度阈值,小于设定值会被抛弃
● –paired:对于双端测序结果,一对 reads 中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准
● –retain_unpaired:对于双端测序结果,一对 reads 中,如果一个 read 达到标准,但是对应的另一个要被抛弃,达到标准的 read 会被单独保存为一个文件
● –gzip 和–dont_gzip:清洗后的数据 zip 打包或者不打包
● –output_dir:输入目录。需要提前建立目录,否则运行会报错
● – trim-n :移除 read 一端的 reads过滤低质量序列
# 使用一个批处理对所有数据进行处理:
mkdir clean
cat ./SRR_Acc_List.txt | while read id; do
trim_galore \
-q 20 \
--length 36 \
--max_n 3 \
--stringency 3 \
--fastqc \
--paired \
-o ./clean/ \
./fastqgz/${id}_1.fastq.gz ./fastqgz/${id}_2.fastq.gz
done完成后文件会输出到 clean 文件夹
十.使用 BWA 将测序数据比对到参考基因组
bwa index [ –p prefix ] [ –a algoType ] <in.db.fasta>
● -P STR:输出数据库的前缀;默认和输入的文件名一致,输出的数据库在其输入文件所在的文件夹,并以该文件名为前缀
● -a [is|bwtsw]:构建 index 的算法,有两个算法:is 是默认的算法,虽然相对较快,但是需要较大的内存,当构建的数据库大于 2GB 的时候就不能正常工作了;bwtsw 对于短的参考序列式不工作的,必须要大于等于 10MB, 但能用于较大的基因组数据,比如人的全基因组bwa mem [options] ref.fa reads.fq [mates.fq]
● -t INT:线程数,默认是 1
● -M:将 shorter split hits 标记为次优,以兼容 Picard’s markDuplicates 软件
● -p:若无此参数:输入文件只有 1 个,则进行单端比对;若输入文件有 2 个,则作为 paired reads 进行比对。若加入此参数:则仅以第 1 个文件作为输入(输入的文件若有 2 个,则忽略之),该文件必须是 read1.fq 和 read2.fa 进行 reads 交叉的数据
● -R STR:完整的 read group 的头部,可以用 ‘\t’ 作为分隔符, 在输出的 SAM 文件中被解释为制表符 TAB. read group 的 ID,会被添加到输出文件的每一个 read 的头部
● -T INT:当比对的分值比 INT 小时,不输出该比对结果,这个参数只影响输出的结果,不影响比对的过程
● -a:将所有的比对结果都输出,包括 single-end 和 unpaired paired-end 的 reads,但是这些比对的结果会被标记为次优对比,使用前面下载的水稻基因组建立索引:
bwa index -a bwtsw Oryza_sativa.IRGSP-1.0.dna.toplevel.fa Oryza_sativa批量进行比对:
cat ./SRR_Acc_List.txt | while read id;
do
bwa mem -v 3 -t 4 ./Oryza_sativa/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa ./clean/${id}_1.fastq.gz ./clean/${id}_2.fastq.gz -o ./compared/${id}.sam
done比对后的结果会输出到 compared 文件夹。
十一.使用 Samtools 对比对结果进行排序和压缩
这个就很常见了,几乎是组学分析必用的软件,之前也详细介绍过了,不再赘述,使用下列批处理将所有数据排序压缩并输出到 sorted 文件夹:
cat ./SRR_Acc_List.txt | while read id ;do
samtools \
sort \
-n -@ 5 \
./compared/${id}.sam \
-o ./sorted/${id}.bam
done接着需要给每个 bam 文件创建索引,后面会用到:
cat ./SRR_Acc_List.txt | while read id;
do
samtools index \
-@ 4 \
./sorted/${id}.bam
done十二.macs2 进行Peak Calling
水稻基因组大小是 3.6e+8,这个基因组的大小是很关键的,如果指定错误或者未指定都会影响后面的分析。使用下列批处理进行分析:
cat ./SRR_Acc_List.txt | while read id;do
macs2 \
callpeak \
-t ./sorted/${id}.bam \
-f BAM \
-g 3.6e+8 \
-n ./peaks/${id} \
done完成后每个样本会输出几个文件:
- NAME_model.r:可视化双峰模型的 R 代码,对双端测序而言,它本身测的就是文库的两端,因此不用建立模型和偏倚,我们只需要对 MACS 设置参数 –nodel 就能略过双峰模型建立的过程
- NAME_peaks.narrowPeak:BED 6+4 格式
- NAME_peaks.xls:包含 peak 信息的 xls 文件,注意 chrStart 是 1-based
- NAME_peaks_summits.bed:BED 格式,包含 peak 的 summits 位置,第 5 列是-log10pvalue,如果想找 motif,推荐使用此文件
十二.deeotools计算基因组段reads覆盖度
deeptools 包含了很多的工具:
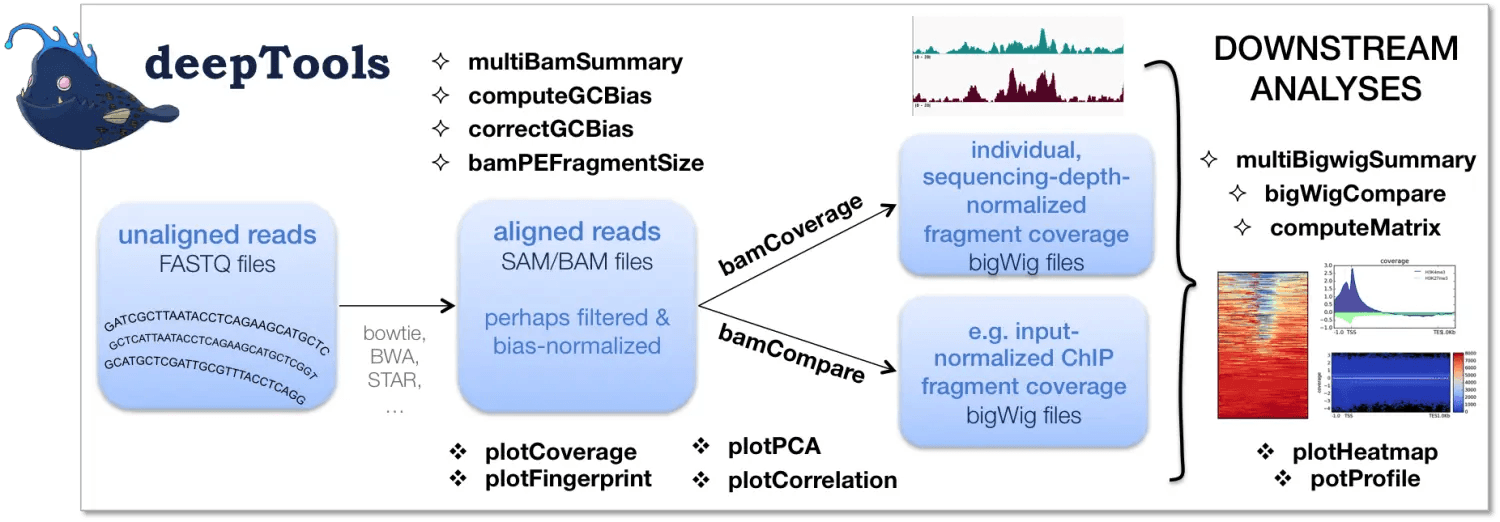
这里我们主要使用的是 bamCoverage 和 computeMatrix。
十三.bamCoverage
bamCoverage 利用测序数据比对结果转换为基因组区域 reads 覆盖度结果。可以自行设定覆盖度计算的窗口大小 (bin)。
bamCoverage -b reads.bam -o coverage.bw
好的,以下是对 deeptools 软件中一些关键参数的详细说明和优化文案,帮助用户更好地理解和使用这些参数。
使用 deeptools 进行基因组区段 reads 覆盖度分析
在进行基因组区段的 reads 覆盖度分析时,deeptools 提供了多种参数来调整和优化你的分析结果。以下是一些必须和推荐使用的参数:
必须的参数
–bam或-b:指定要处理的 BAM 文件。这是进行覆盖度分析的基础输入文件。
bash
deeptools plotFingerprint -b <BAM_FILE> ...–outFileName或-o:设置输出文件的名称。这将决定输出文件的名称和位置。
bash
deeptools plotFingerprint -o <OUTPUT_FILE_NAME> ...–outFileFormat或--of:选择输出文件的格式。支持的格式包括 bigwig 和 bedgraph,根据你的需求选择合适的格式。
bash
deeptools plotFingerprint --outFileFormat bigwig ...标准化参数
必须的参数:
- –bam, -b:要处理的 BAM 文件
- –outFileName, -o:输出文件名
- –outFileFormat, -of:输出文件的格式,可以是 bigwig、bedgraph
deeptools 允许你选择不同的标准化方法来调整覆盖度数据,以下是几种常用的标准化选项:
–normalizeUsing:选择一个标准化方法,可选的参数包括 RPKM、CPM、BPM、RPGC 或 None(默认值)。
-
- RPKM:每千碱基每百万映射 reads 数。公式为: RPKM (per bin)=number of reads per binnumber of mapped reads (in millions)×bin length (kb)RPKM (per bin)=number of mapped reads (in millions)×bin length (kb)number of reads per bin
bash
deeptools plotFingerprint --normalizeUsing RPKM ...-
- CPM:每百万映射 reads 数。公式为: CPM (per bin)=number of reads per binnumber of mapped reads (in millions)CPM (per bin)=number of mapped reads (in millions)number of reads per bin
bash
deeptools plotFingerprint --normalizeUsing CPM ...-
- BPM:每百万映射 reads 数的 bin,类似于 RNA-seq 中的 TPM。公式为: BPM (per bin)=number of reads per binsum of all reads per bin (in millions)BPM (per bin)=sum of all reads per bin (in millions)number of reads per bin
bash
deeptools plotFingerprint --normalizeUsing BPM ...-
- RPGC:基因组内容的 reads 数(1x 归一化)。公式为: RPGC=number of reads per binscaling factor for 1x average coverageRPGC=scaling factor for 1x average coveragenumber of reads per bin
bash
deeptools plotFingerprint --normalizeUsing RPGC ...-
- None:默认值,相当于不进行任何标准化处理。
bash
deeptools plotFingerprint --normalizeUsing None ...此处我使用的命令是:
cat ./SRR_Acc_List.txt | while read id;do
bamCoverage \
-b ./sorted/${id}.bam \
-o ./bigwig/${id}.bw \
--normalizeUsing RPKM
done这里就需要我们前面给 bam 文件创建的 samtools 索引,不然的话会报错。完成之后会输出一系列 bw 文件,这个文件可以导入到 IGV 进行可视化:
十四.computeMatrix
computeMatrix [-h] [--version] ...- reference-point:单个输入文件模式
- scale-regions:多个输入文件模式
必须的参数:
- –regionsFileName, -R:文件名或名称,采用 BED 或 GTF 格式,包含要绘制的区域,如果给出多个床文件,则每个床文件都被视为可以单独绘制的组
- –scoreFileName, -S:bigWig 文件包含要绘制的分数,多个文件应以空格分隔,BigWig 文件可以使用 bamCoverage 或 bamCompare 工具获取
输出参数:
- –outFileName, -out, -o:用于保存“plotHeatmap”和“plotProfile”工具所需的 gzip 压缩矩阵文件的文件名
- –outFileNameMatrix:指定热图矩阵的名称
- –outFileSortedRegions:跳过零或最小/最大阈值后保存区域的文件名,文件中区域的顺序遵循所选的排序顺序
此处我对单个文件进行批处理计算:
cat ./SRR_Acc_List.txt | while read id;do
computeMatrix reference-point \
--referencePoint TSS \
--missingDataAsZero \
-b 3000 -a 3000 \
-R ${id}.bed \
-S ${id}.bw \
-o ${id}.TSS.gz
done- –referencePoint TSS:指定参考点为转录起始位点 (TSS)
- –missingDataAsZero:如果数据缺失,则将其视为零处理
- -b 3000 -a 3000:定义参考点附近的区域大小,这里设置为上游和下游各 3000 个碱基
- -R:指定参考文件,${id} 是当前循环中的 ID
- -S:指定测序数据文件,${id}.bw 是当前循环中的 ID 对应的 bigWig 格式的文件
- -o:指定输出文件名,${id}.TSS.gz 是当前循环中的 ID 作为前缀的输出文件名
绘制热图与折线图:
cat ./SRR_Acc_List.txt | while read id;do
plotHeatmap -m ${id}.TSS.gz -out ${id}.TSS.png
done再同时对多个样本进行计算,将输出结果放在一个文件:
computeMatrix reference-point \
--referencePoint TSS \
--missingDataAsZero \
-b 3000 -a 3000 \
-R SRR28122456.bed \
-S SRR28122455.bw SRR28122456.bw \
-o TSS.gz计算完成之后绘制多个样本的折线图在同一个图内:
plotProfile --dpi 720 -m TSS.gz -out TSS.pdf --plotFileFormat pdf --perGroup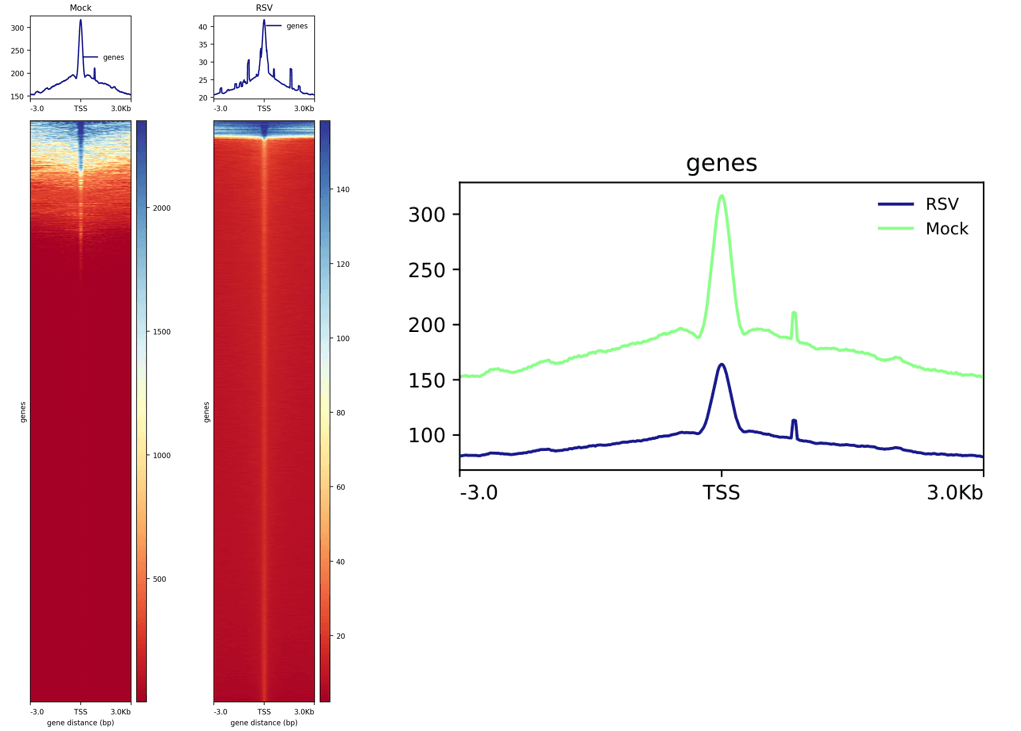
这个图左边是前面绘制的两个热图+折线图,右边是前面绘制多个样本折线图在同一图内。
十五.Chipseek可视化peak
ChIPseeker 是一个功能强大的 R 包,主要用于对 ChIP-seq 数据进行注释和可视化。它不仅可以处理 ChIP-seq 数据,还适用于其他类型的富集数据,如 ATAC-seq、DNase-seq 等。此外,它还支持对长链非编码 RNA(lincRNAs)进行注释。以下是关于如何安装和使用 ChIPseeker 的详细说明。
安装 ChIPseeker 包
要在 R 环境中安装 ChIPseeker,你可以使用以下步骤:
- 打开 R 环境:
-
- 确保你的 R 环境已经启动。
- 安装 BiocManager:
-
- BiocManager 是一个用于安装生物信息学相关 R 包的工具。如果你还没有安装 BiocManager,可以通过以下命令安装:
r
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")- 安装 ChIPseeker:
-
- 使用 BiocManager 安装 ChIPseeker:
r
BiocManager::install("ChIPseeker")- 加载 ChIPseeker:
-
- 安装完成后,使用以下命令加载 ChIPseeker:
r
library(ChIPseeker)ChIPseeker 的主要功能
- 注释功能:
-
- ChIPseeker 可以对 peak 位置进行注释,识别 peak 附近的基因,并提供详细的注释信息,如基因名称、功能等。
- 可视化功能:
-
- ChIPseeker 的强大之处在于其丰富的可视化功能。它可以生成多种类型的图表,如基因组浏览器轨迹图、热图、条形图等,帮助用户直观地理解数据。
- 适用性广泛:
-
- 虽然最初设计用于 ChIP-seq 数据,但 ChIPseeker 也适用于其他类型的富集数据,如 ATAC-seq、DNase-seq 等。它还可以用于注释长链非编码 RNA(lincRNAs)。
所需的文件有两个,一个是 BED 格式的文件,至少得有染色体名字、染色体起始位点和染色体终止位点,其它信息如 name,score,strand 等可有可无。这里直接用前面 macs2 输出的 bed 文件即可。另一个是有注释信息的 TxDb 对象,Bioconductor 包提供了 30 个 TxDb 包,包含了很多物种,如人,老鼠等。当所研究的物种没有已有的 TxDb 时,可通过 GenomicFeatures 包使用基因组注释文件进行制作:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GenomicFeatures")
BiocManager::install("txdbmaker")这里我使用前面下载的水稻注释文件进行制作:
library("GenomicFeatures")
spombe <- makeTxDbFromGFF("Oryza_sativa.IRGSP-1.0.57.gff3")然后就可以读入 bed 文件进行可视化:
# 为了这里方便分析,将两个 bed 文件按照样本信息进行重命名了
Mock <- readPeakFile('./peaks/Mock.bed')
RSV <- readPeakFile('./peaks/RSV.bed')
Mock_peakAnno <- annotatePeak(Mock, tssRegion =c(-3000, 3000), TxDb = spombe)
RSV_peakAnno <- annotatePeak(RSV, tssRegion =c(-3000, 3000), TxDb = spombe)
plotAnnoPie(Mock_peakAnno)
plotAnnoPie(RSV_peakAnno)也可以将多个样本绘制在同一个图:
peaks <- list(Mock_peakAnno=Mock, RSV_peakAnno=RSV)
peakAnno <- lapply(peaks, annotatePeak, tssRegion = c(-3000, 3000), TxDb = spombe)
plotAnnoBar(peakAnno)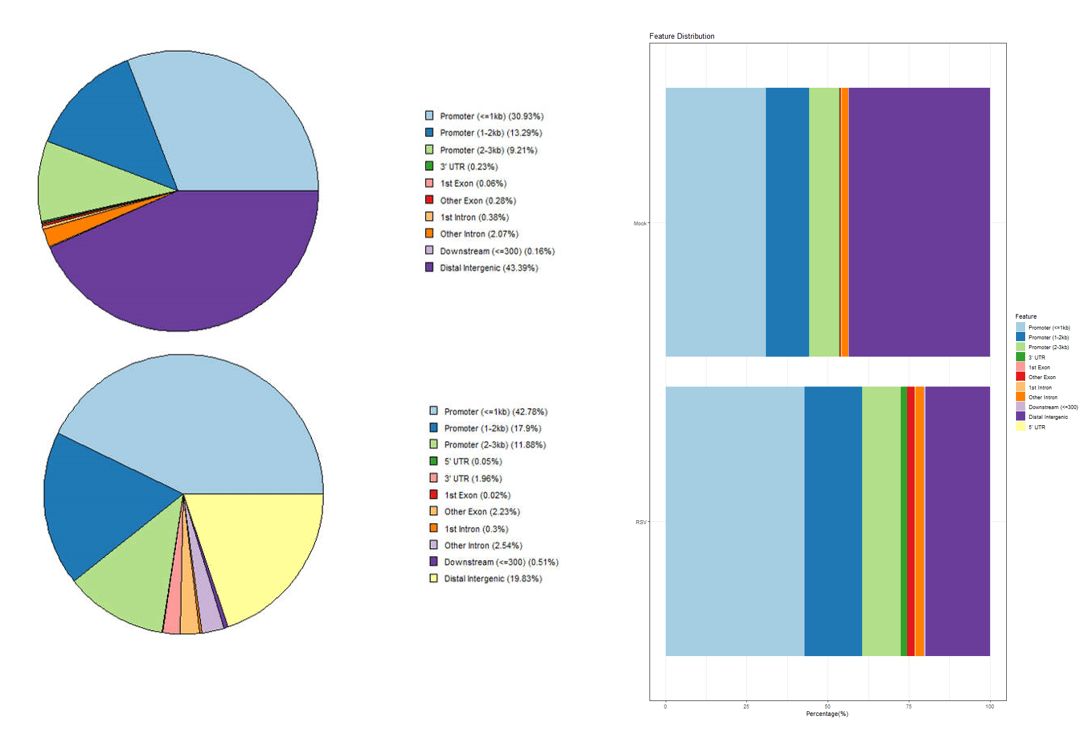
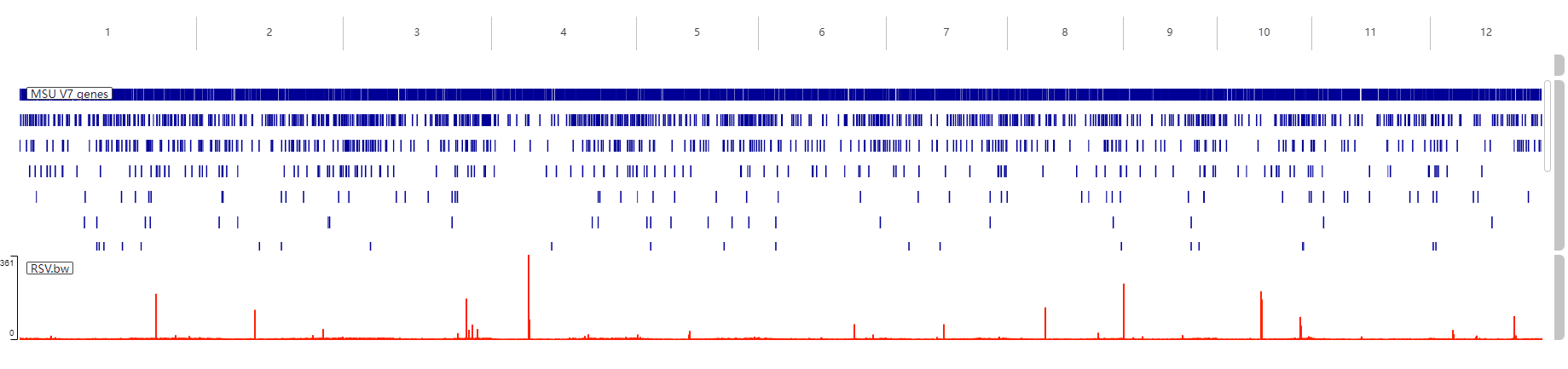
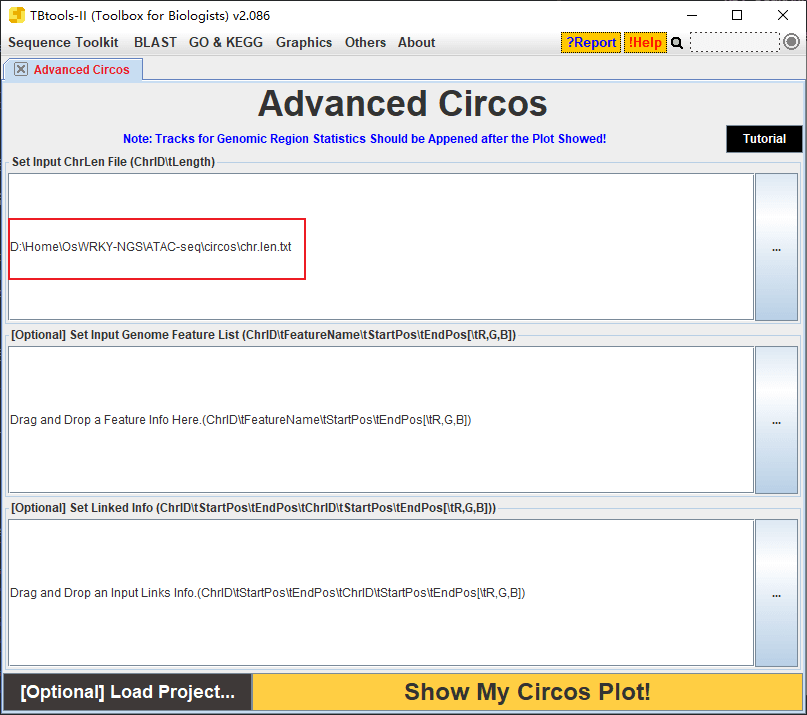
十六.在染色体上可视化 peaks 的位置
这个使用 TBtools 来进行,先制作一个染色体长度文件:
1 43270923
2 35937250
3 36413819
4 35502694
5 29958434
6 31248787
7 29697621
8 28443022
9 23012720
10 23207287
11 29021106
12 27531856然后使用 TBtools 的 circos 绘图工具,只把染色体长度文件导入:
修改 macs2 输出文件中的 bed 文件,只保留染色体、开始位置、终止位置和值四列即可,如:
1 1074 1075 33.7137
1 84253 84254 2.21489
1 104155 104156 5.22729
1 104518 104519 3.93076
1 104876 104877 6.8855将文件导入到 TBtools:
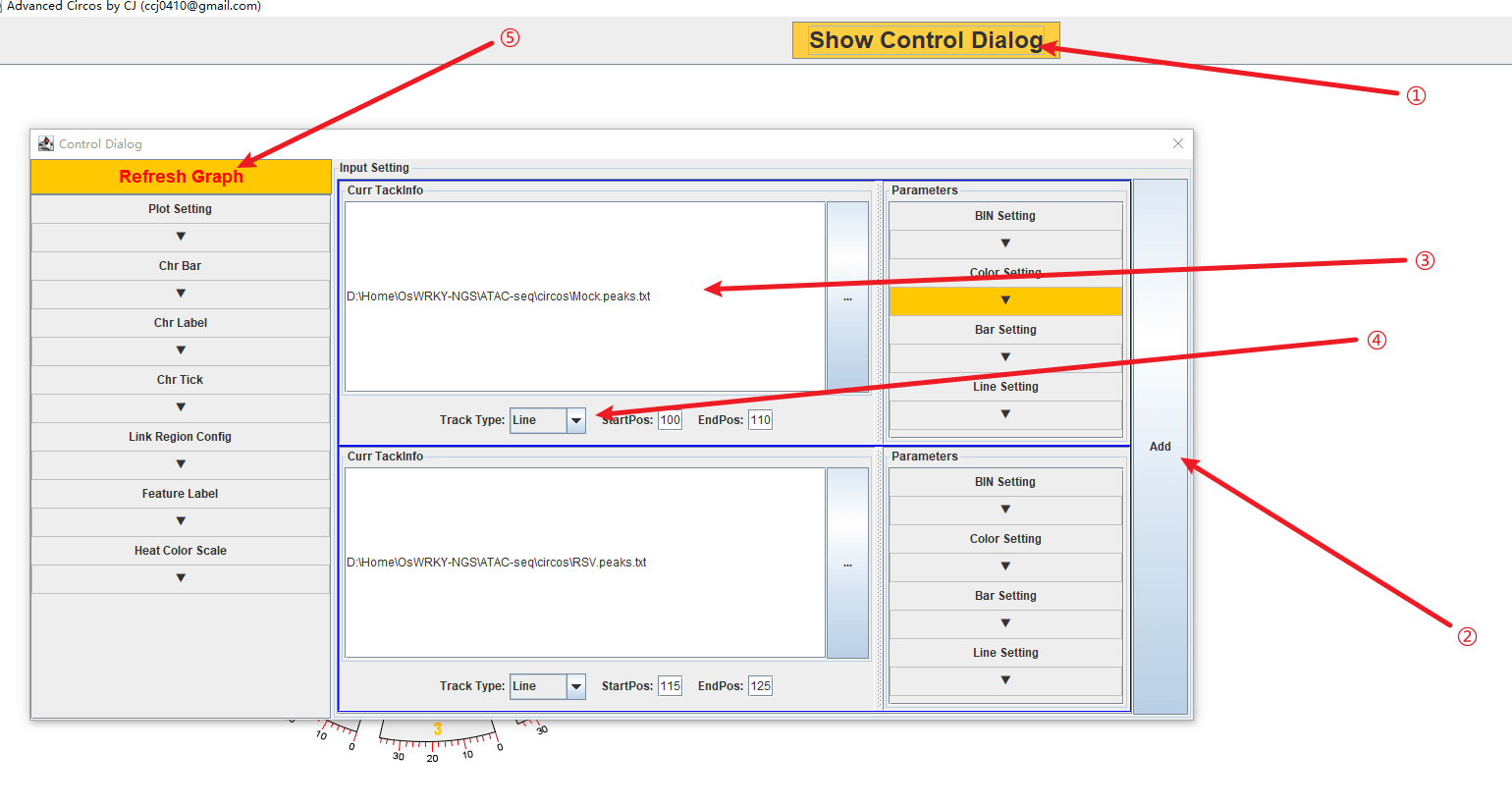
然后改一下颜色、字体什么的进行一下简单的美化:
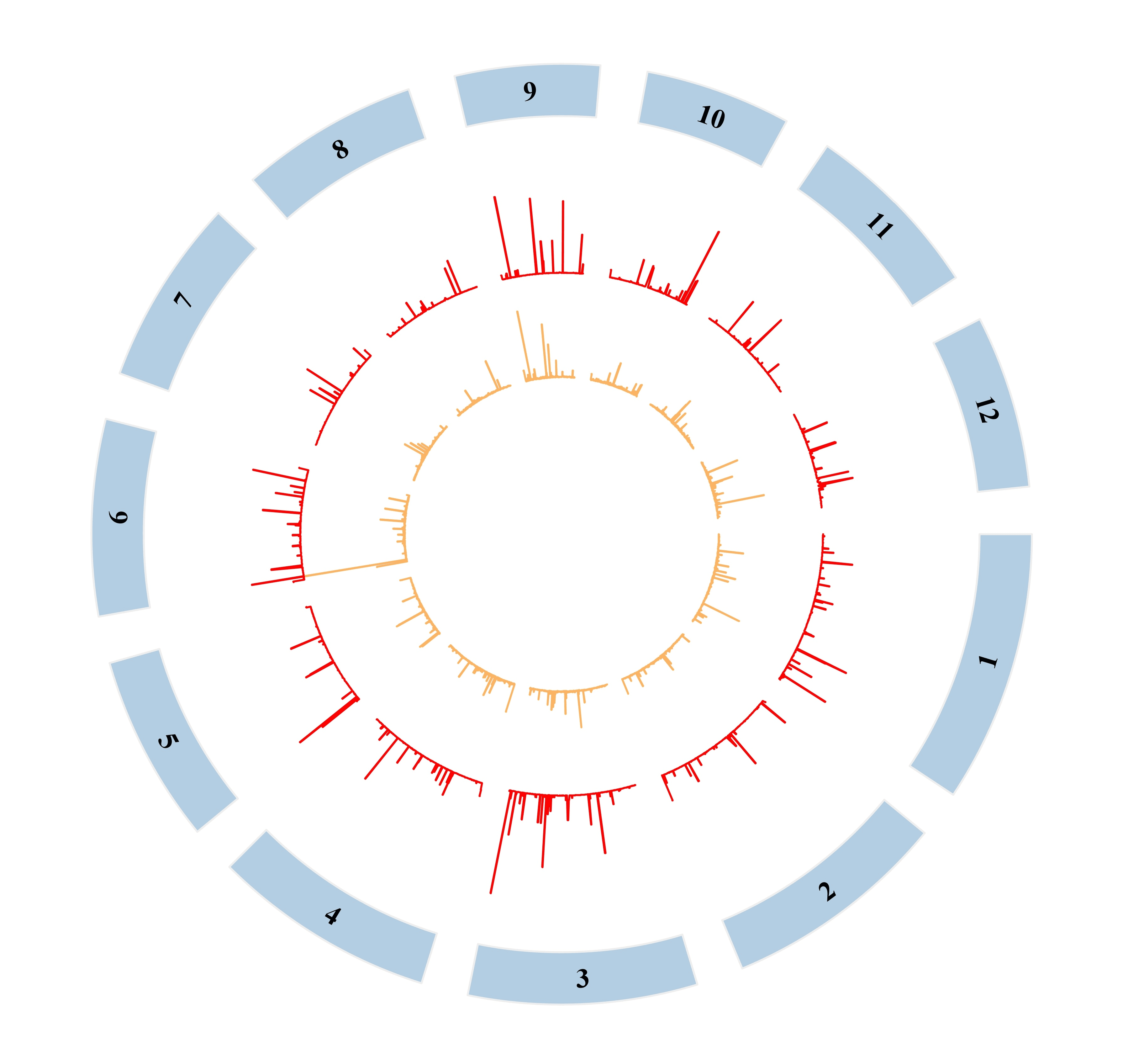
到这里大致的流程基本上就完成了,还有一些分析就是视情况才做的了。















 166
166

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









