







Visium HD为了获取单细胞级数据,前面我们介绍了基于高清的H&E图像使用StarDist进行细胞核分割,然后将覆盖在识别出来的细胞核区域上的2um bin进行合并,将这些合并的bins当做一个细胞进行后续分析。
不管是10x官方提供的细胞核分割方法(https://www.10xgenomics.com/analysis-guides/segmentation-visium-hd), 或是bin2cell (https://github.com/Teichlab/bin2cell), 抑或是上节我们测试的ENACT (https://github.com/Sanofi-Public/enact-pipeline)其本质都是一样的,使用StarDist预训练的2D_versatile_he模型首先识别细胞核轮廓,然后将覆盖在细胞核轮廓上的bins进行合并,无非是bin-to-cell合并的方法不同罢了。
需要特别强调下,只要是使用StarDist预训练的2D_versatile_he模型进行分割的,那一定分割的是细胞核,尽管StarDist也是可以进行细胞轮廓识别,但是2D_versatile_he模型是使用DAPI染的细胞核图像训练的,而上面我们说的3种分割都是用的2D_versatile_he模型,所以记住它们都是细胞和分割,分割后bin-to-cell获取的是接近snRNA-seq结果会丢失细胞质的表达信息。
为了识别出细胞边界,我们可以使用Cellpose (https://www.cellpose.org/)进行单细胞分割,然后再bin-to-cell获得的就是单细胞精度的数据了。
Cellpose提供了多种预训练模型,主要包括2类:
1. 细胞质模型(cyto):适用于细胞质分割,通常用于处理细胞形态较为复杂的图像。
2. 细胞核模型(nuclei):专门用于细胞核的分割,适合于需要高精度核定位的应用。
Cellpose在Visium HD数据细胞分割中的优势:
高分辨率细胞分割:
Cellpose作为一种深度学习模型,能够有效地处理这些高分辨率图像,提供精确的细胞分割,从而确保后续分析的准确性;
自动化处理:
Cellpose能够自动识别和分割图像中的细胞,减少了人工标注的需求;
适应性强:
Cellpose经过多样化的数据集训练,具有较强的通用性,能够适应不同类型的细胞图像,而无需用户进行大量的重新训练或调整参数;
支持复杂组织结构:
Cellpose能够有效处理复杂且细胞密度较高且复杂的组织结构,能准确识别重叠细胞和多样化形态,提高分割精度。
以下是使用Cellpose进行细胞分割,然后将2um bin基因表达合并得到单细胞精度表达数据,代码我都进行了详细注释。
# 导入必要的库
import numpy as np
import pandas as pd
import scanpy as sc
import tifffile
import geopandas as gpd
from shapely.geometry import Point
from cellpose import models, core
# 定义读取图像的函数,并可选择下采样因子
def read_dapi_image(img_path: str, downscale_factor: int = 2) -> np.ndarray:
img = io.imread(img_path) # 读取图像文件
print(img.shape) # 打印图像的形状
return downscale_image(img, downscale_factor=downscale_factor) # 返回下采样后的图像
# 定义下采样图像的函数
def downscale_image(img: np.ndarray, downscale_factor: int = 2) -> np.ndarray:
# 计算每个轴上所需的填充量
pad_height = (downscale_factor - img.shape[0] % downscale_factor) % downscale_factor
pad_width = (downscale_factor - img.shape[1] % downscale_factor) % downscale_factor
new_shape = (img.shape[0] + pad_height, img.shape[1] + pad_width, img.shape[2]) # 新图像形状
new_image = np.zeros(new_shape, dtype=img.dtype) # 创建新的零填充图像
for channel in range(img.shape[2]):
new_image[:, :, channel] = np.pad(
img[:, :, channel], ((0, pad_height), (0, pad_width)), mode="constant"
) # 对每个通道进行零填充
return new_image # 返回填充后的图像
# 定义运行Cellpose进行细胞分割的函数
def run_cellpose(img: np.ndarray, model_path: str):
use_GPU = core.use_gpu() # 检查是否使用GPU
model = models.CellposeModel(gpu=use_GPU, pretrained_model=model_path) # 加载Cellpose模型
channels = [0, 0] # 设置通道(这里假设为单通道)
masks, flows, styles = model.eval(
[img],
channels=channels,
diameter=model.diam_labels,
flow_threshold=1,
cellprob_threshold=0,
batch_size=4,
) # 执行细胞分割并返回掩膜、流和样式信息
return (masks, flows, styles) # 返回分割结果
# 设置Visium HD数据的文件路径并读取图像
img_path = "./data/visiumHD_colorectal_cancer/P1_CRC/Visium_HD_Human_Colon_Cancer_P1_tissue_image.btf"
maxed_visium = read_dapi_image(img_path, downscale_factor=1) # 读取并下采样图像
# 设置分块参数和模型路径
chunk_per_axis = 2 # 每个轴上分块数目
mp = 'cyto' # 使用细胞质模型进行分割
# 对图像进行分块并调用Cellpose进行细胞分割
masks_top_left, flows, styles = run_cellpose(
maxed_visium[
: np.shape(maxed_visium)[0] // chunk_per_axis,
: np.shape(maxed_visium)[1] // chunk_per_axis,
:,
],
model_path=mp,
)
masks_top_right, flows, styles = run_cellpose(
maxed_visium[
: np.shape(maxed_visium)[0] // chunk_per_axis,
np.shape(maxed_visium)[1] // chunk_per_axis :,
:,
],
model_path=mp,
)
masks_bottom_left, flows, styles = run_cellpose(
maxed_visium[
np.shape(maxed_visium)[0] // chunk_per_axis :,
: np.shape(maxed_visium)[1] // chunk_per_axis,
:,
],
model_path=mp,
)
masks_bottom_right, flows, styles = run_cellpose(
maxed_visium[
np.shape(maxed_visium)[0] // chunk_per_axis :,
np.shape(maxed_visium)[1] // chunk_per_axis :,
:,
],
model_path=mp,
)
# 初始化全掩膜数组,准备合并分块结果
constant = 1000000 # 用于区分不同块的常量值
full_mask = np.zeros_like(maxed_visium[:, :, 0], dtype=np.uint32) # 创建与原图相同大小的掩膜数组
# 将各个块的掩膜合并到全掩膜中,并加上常量以区分不同区域
full_mask[
: np.shape(maxed_visium)[0] // chunk_per_axis,
: np.shape(maxed_visium)[1] // chunk_per_axis,
] = masks_top_left[0]
full_mask[
: np.shape(maxed_visium)[0] // chunk_per_axis,
np.shape(maxed_visium)[1] // chunk_per_axis :,
] = masks_top_right[0] + (constant)
full_mask[
np.shape(maxed_visium)[0] // chunk_per_axis :,
: np.shape(maxed_visium)[1] // chunk_per_axis,
] = masks_bottom_left[0] + (constant * 2)
full_mask[
np.shape(maxed_visium)[0] // chunk_per_axis :,
np.shape(maxed_visium)[1] // chunk_per_axis :,
] = masks_bottom_right[0] + (constant * 3)
# 将全掩膜中值为常量的部分设置为0,表示没有细胞
full_mask = np.where(full_mask % constant == 0, 0, full_mask)
# 将全掩膜保存为PNG格式文件
tifffile.imsave("./visium_hd/segmentation.png", full_mask)
# 设置Visium HD数据目录路径以加载空间转录组数据
dir_base = "./data/visiumHD_colorectal_cancer/P1_CRC/outs/binned_outputs/square_002um/"
# 加载Visium HD数据中的基因表达矩阵(HDF5格式)
raw_h5_file = dir_base + "filtered_feature_bc_matrix.h5"
adata = sc.read_10x_h5(raw_h5_file) # 使用Scanpy读取数据
# 加载组织位置坐标文件(Parquet格式)
tissue_position_file = dir_base + "spatial/tissue_positions.parquet"
df_tissue_positions = pd.read_parquet(tissue_position_file)
# 将数据框的索引设置为条形码,以便后续合并操作使用
df_tissue_positions = df_tissue_positions.set_index("barcode")
# 在数据框中创建索引以检查合并情况
df_tissue_positions["index"] = df_tissue_positions.index
# 将组织位置添加到AnnData对象的元数据中(obs)
adata.obs = pd.merge(adata.obs, df_tissue_positions, left_index=True, right_index=True)
# 从坐标数据框创建GeoDataFrame,以便进行空间分析
geometry = [
Point(xy)
for xy in zip(
df_tissue_positions["pxl_col_in_fullres"],
df_tissue_positions["pxl_row_in_fullres"],
)
]
gdf_coordinates = gpd.GeoDataFrame(df_tissue_positions, geometry=geometry)
## 将捕获区域分配给单个细胞
# 检查点掩膜是否在全掩膜范围内,以确保坐标有效性
point_mask = (
gdf_coordinates["pxl_row_in_fullres"].values.astype(int) < np.shape(full_mask)[0]
) & (gdf_coordinates["pxl_col_in_fullres"].values.astype(int) < np.shape(full_mask)[1])
# 根据有效坐标从全掩膜中提取细胞ID信息
cells = full_mask[
gdf_coordinates["pxl_row_in_fullres"].values.astype(int)[point_mask],
gdf_coordinates["pxl_col_in_fullres"].values.astype(int)[point_mask],
]
gdf_coordinates = gdf_coordinates[point_mask]
gdf_coordinates["cells"] = cells # 将细胞ID添加到GeoDataFrame中
assigned_regions = gdf_coordinates[gdf_coordinates["cells"] > 0]
# 将细胞ID信息合并到AnnData对象中,以便后续分析使用
adata.obs = adata.obs.merge(
assigned_regions[["cells"]], left_index=True, right_index=True, how="left"
)
adata = adata[~pd.isna(adata.obs["cells"])] # 移除没有细胞ID的数据行
adata.obs["cells"] = adata.obs["cells"].astype(int) # 将细胞ID转换为整数类型
## 在捕获区域内汇总转录本计数
from scipy import sparse
# 按唯一细胞ID对数据进行分组,以便汇总转录本计数
groupby_object = adata.obs.groupby(["cells"], observed=True)
counts = adata.X # 提取基因表达计数矩阵
spatial_coords = adata.obs[["pxl_row_in_fullres", "pxl_col_in_fullres"]].values
N_groups = groupby_object.ngroups # 获取唯一细胞ID数量(组数)
N_genes = counts.shape[1] # 获取基因数量
# 初始化稀疏矩阵以存储每个细胞的基因计数汇总结果
summed_counts = sparse.lil_matrix((N_groups, N_genes))
polygon_id = [] # 存储多边形ID列表
cell_coords = [] # 存储细胞坐标列表
row = 0
# 遍历每个唯一多边形,计算基因计数的总和。
for polygons, idx_ in groupby_object.indices.items():
summed_counts[row] = counts[idx_].sum(0) # 汇总该多边形内所有细胞的基因计数
cell_coords.append(np.mean(spatial_coords[idx_], axis=0)) # 获取该多边形内细胞坐标均值作为代表坐标
row += 1
polygon_id.append(polygons) # 添加多边形ID到列表中
cell_coords = np.array(cell_coords)
summed_counts = summed_counts.tocsr() # 转换为压缩稀疏行矩阵格式
# 创建一个新的AnnData对象,用于存储汇总后的基因计数矩阵及其元数据。
grouped_adata = anndata.AnnData(
X=summed_counts,
obs=pd.DataFrame(polygon_id, columns=["cells"], index=polygon_id),
var=adata.var,
)
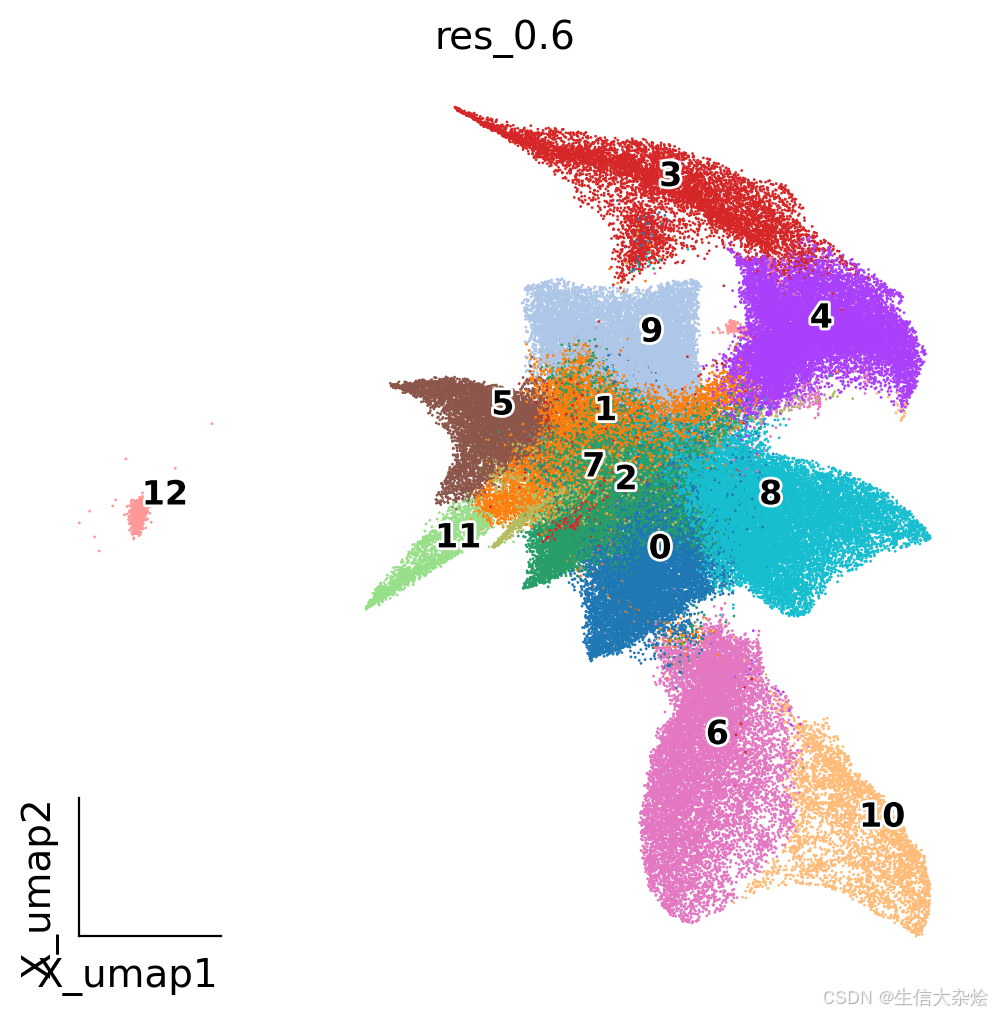
grouped_adata.write('adata_cellpose.h5ad')拿到细胞分割后的结果后,降维聚类看下结果,对比之前合并8um bin结果看起来,大群基本上分开了,效果挺好。
题外话
其实我们还可以使用Qupath导入H&E图像,使用Cellpose或者一些开源的机器学习算法可视化进行细胞分割,好处是可以自己训练模型,然后细胞分割后直观就能看到,如果不满意就可以微调参数进行分割,直到能将绝大多数细胞分割出来,最后可以导出这些细胞的坐标信息,然后就是使用上面bin-to-cell部分的代码,就能达到更好的结果。
微信公众号: 生信大杂烩


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









