







前几天有同学后台私信我一篇2024年10月份在bioRxiv预印的文章ENACT: End-to-End Analysis of Visium High Definition (HD) Data, 希望我帮助解读和测试使用看看效果怎样。我大致看了遍文章,然后使用10x Visium HD的结直肠癌样本数据测试了一遍,这里做个记录,供大家参考。
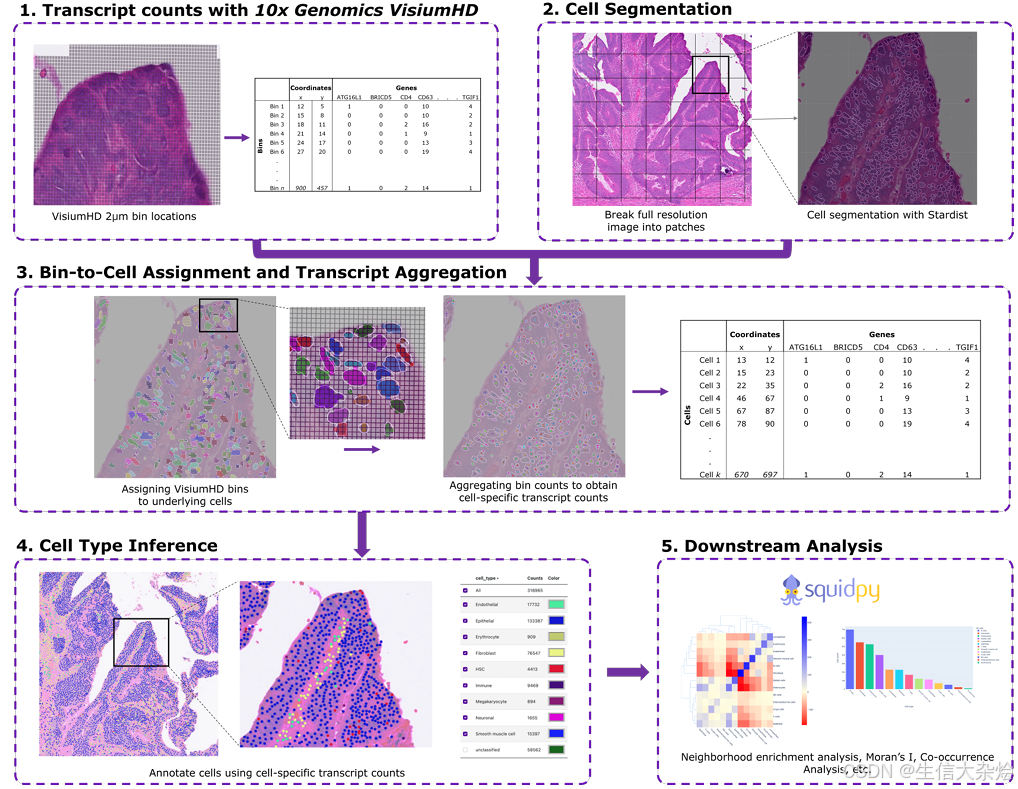
根据文章中提供的信息, ENACT的处理和分析流程主要包括以下几个步骤:
1.使用Stardist对全分辨率的组织图像进行细胞分割, 预测细胞边界(其实就是细胞核分割,因为他使用的是Stardist的2D_versatile_he模型进行分割,和之前我们介绍发果的10x官方提供的细胞核分割方法一样);
2.将每个细胞边界范围内覆盖的2µmx2µm bin进行合并, 得到一个2D的细胞-基因矩阵;
3.使用Sargent、CellAssign或CellTypist等方法,根据细胞的转录本计数来推断细胞类型;
4.将预测的细胞类型标签和它们在组织中的空间位置封装到AnnData对象中,可以使用Squidpy进行后续的空间统计分析,如邻域富集、Moran's I和共现分析等;
总之,ENACT提供了一个集成了先进的深度学习细胞分割模型和多种bin-to-cell分配策略的全面处理流程,能够从Visium HD数据中准确地提取单细胞转录本信息,并进行后续的空间分析。
一句话总结,其实它的原理和bin2cell一样,基于HE图像首先识别出细胞核轮廓,就是多了提供了4种bin-to-cell分配方法,然后得到单细胞核数据,使用流程内置的几种自动注释方法就能根据细胞内的转录本计数来推断细胞类型,最后可以使用Squidpy进行后续的空间统计分析。
ENACT提供了4种将转录本计数映射到单个细胞的bin-to-cell分配方法(我认为这可能是唯一新颖的地方):
1.朴素方法(Naive):类似于Bin2cell方法只考虑唯一bins,删除细胞重叠区域的bins;
2.按面积加权(Weighted-by-Area):根据bins与细胞的重叠面积比例来分配转录本计数, 这种方法可以将所有重叠的bin中的转录本全部分配, 但忽略了相邻细胞的差异;
3.按转录本加权(Weighted-by-Transcript):考虑相邻细胞的基因表达情况, 根据重叠细胞的归一化转录本计数来加权分配bin中的转录本。这种方法可以更准确地分配转录本,但如果bin中包含在重叠细胞中未表达的基因,则会导致信息丢失;
4.按Cluster加权(Weighted-by-Cluster):首先使用朴素方法获得初步的单细胞转录本估计,然后使用K-means聚类将细胞分组,并利用每个簇的平均转录本计数来分配bin中的转录本。这种方法可以解决"按转录本加权"方法中的信息丢失问题。
文章中构造了几种不同数据,证明了在不同情况下,选择合适的bin-to-cell分配方法很重要。总结起来各个方法的特点如下:
朴素方法简单快速,但准确性较低;
按面积加权可以全面分配转录本,但忽略了细胞差异;
按转录本加权考虑了细胞差异,但可能会丢失信息;
按簇加权则可以更好地平衡准确性和效率。
分析代码
import scanpy as sc
from enact.pipeline import ENACT
so_hd = ENACT(
cache_dir="./cache", # 输出结果文件位置
wsi_path="/data/visiumHD_colorectal_cancer/P1_CRC/Visium_HD_Human_Colon_Cancer_P1_tissue_image.btf", # 高清HE图像位置
visiumhd_h5_path="/data/visiumHD_colorectal_cancer/P1_CRC/outs/binned_outputs/square_002um/filtered_feature_bc_matrix.h5", # 2um bin基因表达
tissue_positions_path="/data/visiumHD_colorectal_cancer/P1_CRC/outs/binned_outputs/square_002um/spatial/tissue_positions.parquet", # 2um bin坐标
analysis_name="demo-colon", # 会在cache_dir文件夹下生成analysis_name文件夹存放结果文件
seg_method="stardist", # 细胞分割方法,现只支持stardist方法
bin_to_cell_method="weighted_by_area", # 设置bin_to_cell分配方法
cell_annotation_method="celltypist", # 使用celltypist对识别出来的单细胞进行注释
cell_typist_model="Human_Colorectal_Cancer.pkl", # celltypist注释参考数据
segmentation=True, # 进行细胞分割
bin_to_geodataframes=True, # convert bin to geodataframes
bin_to_cell_assignment=True, # assign cells to bins
cell_type_annotation=True, # run cell type annotation
use_hvg=False #这里我为了拿到adata包含所有基因表达,就没有取高变基因
)
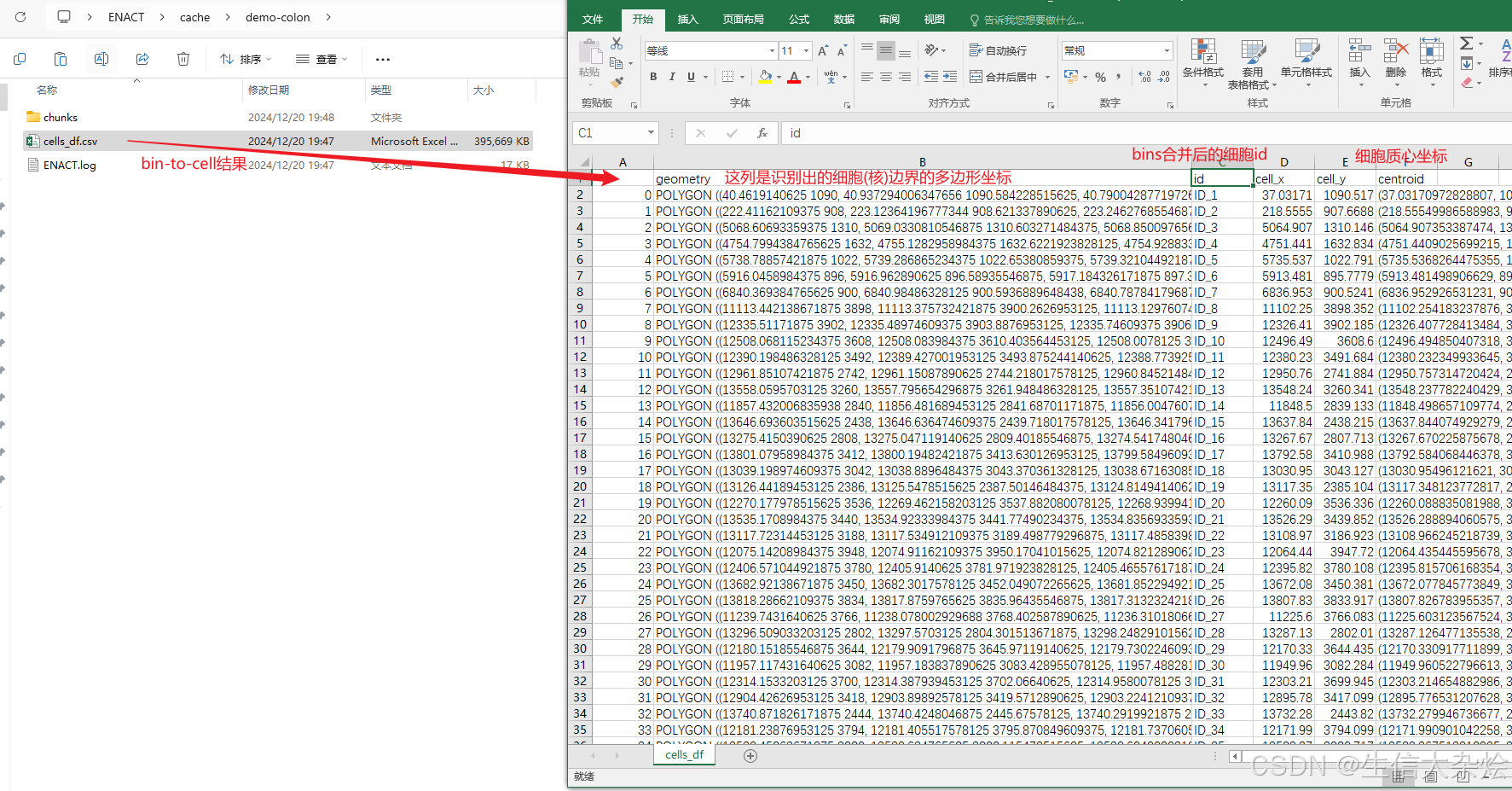
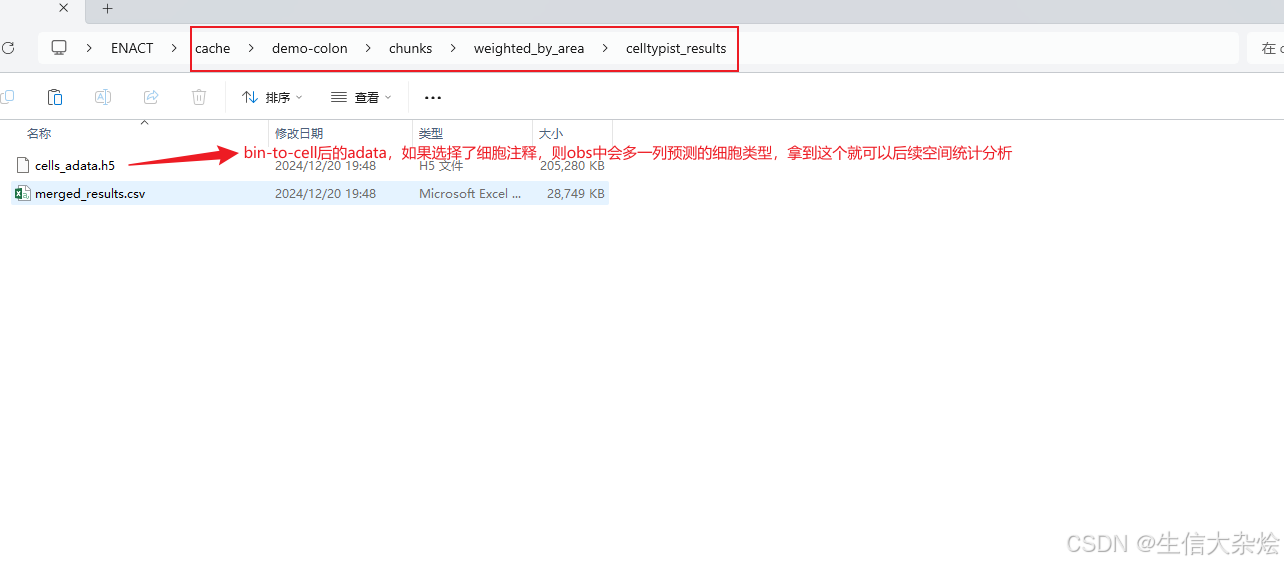
so_hd.run_enact()输出结果
总结
我们使用bin_to_cell_method="weighted_by_area"进行分析,在P1_CRC样本识别到了大概30万个细胞,拿到adata后,我们首先过滤低质量细胞,然后进行降维聚类(如果bin-to-cell结果准确的话,降维聚类后的UMAP应该像单细胞测序结果一样),但是发现UMAP结果还是很混杂,并且比单纯细胞核分割结果还差,猜测可能我们用的这张片子细胞分割是不是用其他的bin_to_cell方法会更好。
新的想法
不管是10x官方的细胞核分割,还是bin2cell,抑或是今天说的ENACT方法其实都是为了一个目的,就是将2um bin数据按照细胞进行合并,获得准确的单细胞分辨率的数据,但是上述的几个方法却都是使用stardist在HE图像上识别细胞核,无非是bin-to-cell的分配方法不同。既然要分割细胞,而且图像是HE,那为什么不使用Cellpose检测和分割细胞整体边界,然后将bins合并到细胞呢,下节我们尝试使用Cellpose细胞分割,然后结合ENACT的bin-to-cell各种方法,获取单细胞表达数据,看看效果怎样。
微信公众号:生信大杂烩

















 611
611

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









