






记录下最近阅读的一篇于2022年8月在Nature Communications上发表的文章"scRNA-seq of gastric tumor shows complex intercellular interaction with an alternative T cell exhaustion trajectory",记录下翻译过程,以备后期随时翻阅。

摘要
胃癌(GC)的肿瘤微环境(TME)对于肿瘤控制被认为很重要,但 GC 的具体特征尚未完全理解。我们生成了来自 10 位 GC 患者的匹配肿瘤周围组织和血液的 166,533 个细胞的图谱。我们的结果显示,肿瘤相关的基质细胞(TASCs)上调了 Wnt 信号和血管生成的活性,并与生存率负相关。肿瘤相关巨噬细胞和 LAMP3+ 树突细胞参与介导 T 细胞活性,并与 TASCs 形成细胞间互动枢纽。克隆型和轨迹分析表明,Tc17(IL-17+CD8+ T 细胞)来源于组织驻留记忆 T 细胞,并可随后分化为耗竭 T 细胞,这暗示了 T 细胞耗竭的另一种途径。我们的结果表明,IL17+ 细胞可能通过 IL17、IL22 和 IL26 信号促进肿瘤进展,凸显了以 IL17+ 细胞及相关信号通路为治疗靶点治疗 GC 的可能性。
引言
胃癌(GC),包含许多分子亚型,是全球第五常见的恶性肿瘤,但是癌症相关死亡的第三大原因,2018 年估计有 783,000 人死亡。虽然 GC 在早期原发阶段具有较高的可治疗性,但大多数患者在被检测出时已处于晚期或转移性阶段,预后相对较差。免疫疗法,特别是针对 PD-1 和 CTLA4 的抗体,已经为各种癌症类型的治疗带来了范式转变,如黑色素瘤,但在 GC 中的响应率相对较低。许多先前的研究表明,肿瘤间异质性和细胞组成的个体差异与生存率有关,突出了解剖复杂和动态的肿瘤微环境(TME)生物学特性以利用先进干预措施对抗它的未满足需求。
最近,单细胞 RNA 测序(scRNA-seq)已成功用于解析 GC 的生态系统,以解剖和发现感兴趣的底层肿瘤生物学。例如,Wang 等人和 Zhang 等人指出了原发性和转移性胃腺癌的转录异质性和谱系多样性,并为诊断和预后提供了标志性基因。Zhang 等人和 Yin 等人勾勒了 GC 发生和发展期间广泛的细胞表型重塑,并且还识别了早期 GC 检测的标记物。Kumar 等人显示了弥漫型胃肿瘤中等离子细胞比例的增加,并研究了癌相关成纤维细胞中的 INHBA-FAP 轴。
在这项工作中,我们应用 scRNA-seq 来绘制来自 10 位 GC 患者的肿瘤、相邻正常组织和匹配外周血中的免疫、基质和上皮隔间的转录景观,并结合 T/B 细胞受体(TCR/BCR)库谱分析。我们的结果表明,肿瘤组织中的基质细胞经历了显著的转变,并展示了广泛的促肿瘤特性。细胞 - 细胞通信分析显示,TASCs、TAMs 和 LAMP3+DCs 是 TME 中复杂细胞互动的重要媒介。结合 TCR 克隆型信息和轨迹分析,我们展示了 GC 中的 Tc17 可能源自组织驻留记忆群体,并可随后分化为耗竭状态,这暗示了 T 细胞耗竭的另一条途径。我们的结果表明,IL17+ 细胞和介导 IL17+ 细胞与肿瘤细胞通信的途径是治疗 IL17+ 阳性胃癌的潜在治疗靶点。
结果
胃癌微环境的单细胞 RNA 测序图谱
为了探索参与胃癌(GC)的细胞类型多样性,我们在单细胞分辨率下生成了十位原发性 GC 患者肿瘤组织中所有可行细胞的 scRNA-seq 剖面,这些患者在采样前未接受治疗,以及来自匹配的外周血和相邻正常组织(图 1a 及补充图 1a, b 和补充数据 1)。同时,我们还对同一样本进行了整体外显子测序(WES)和整体 RNA 测序,少数例外除外。
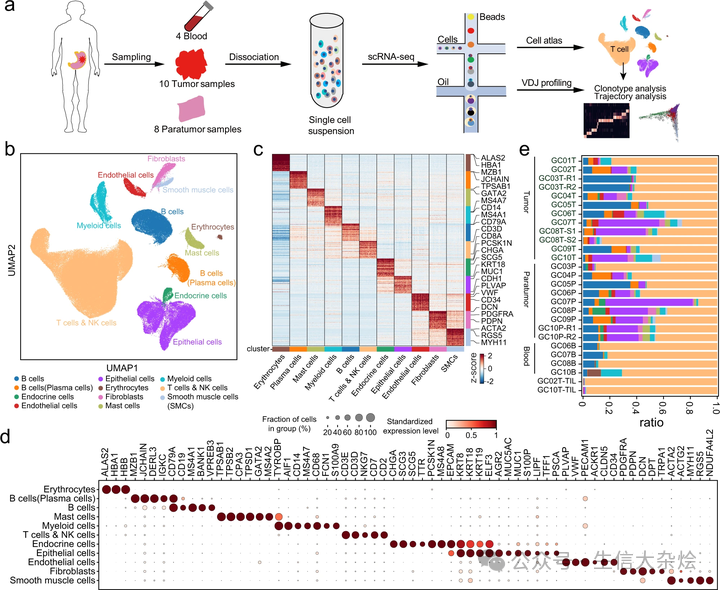
添加图片注释,不超过 140 字(可选)
a. 描述胃癌(GC)、外周血和肿瘤周围细胞样本处理和 scRNA 测序的工作流程以及随后的分析方法。b. 来自 10 位患者的 166,533 个单细胞的统一流形近似和投影(UMAP),按主要细胞类型着色。c. 热图显示主要细胞类型(列)中差异表达基因(行),并标出了典型的标记基因。d. 点图显示图 1b 中簇的标记基因。点的大小表示表达该基因的细胞比例,颜色表示标准化表达水平。e. 每个样本中检测到的细胞类型比例,颜色与图 1b 中相同。GC01-GC10 代表 10 位 GC 患者;B/P/T 分别代表从血液、肿瘤周围和肿瘤组织中分离的细胞;TIL 代表肿瘤浸润淋巴细胞;GC03T-R1/R2 和 GC10P-R1/R2 代表两个技术重复;GC08T-S1/S2 代表同一肿瘤组织的两个不同部位。
GC 中的恶性细胞表现出广泛的异质性
被定义为上皮细胞的细胞被提取出来并重新聚类。为了区分肿瘤细胞和正常细胞,我们首先根据 Zhang 等人描述的方法,基于肿瘤和正常组织的特征基因表达计算肿瘤评分(图 2a)。其次,我们通过 inferCNV 算法,一种利用 scRNA-seq 数据估计基因组拷贝数变化的算法,计算细胞的拷贝数变异(CNV)评分(图 2b 和补充图 2b)。最后,我们通过比较肿瘤组织与肿瘤周围组织的 WES 数据,为每位患者调用特定于肿瘤的突变,然后,在匹配的单细胞数据中搜索这些特定于肿瘤的突变。这样的突变在 4 个上皮细胞簇中被富集(图 2c,补充图 2c,详见方法部分)。
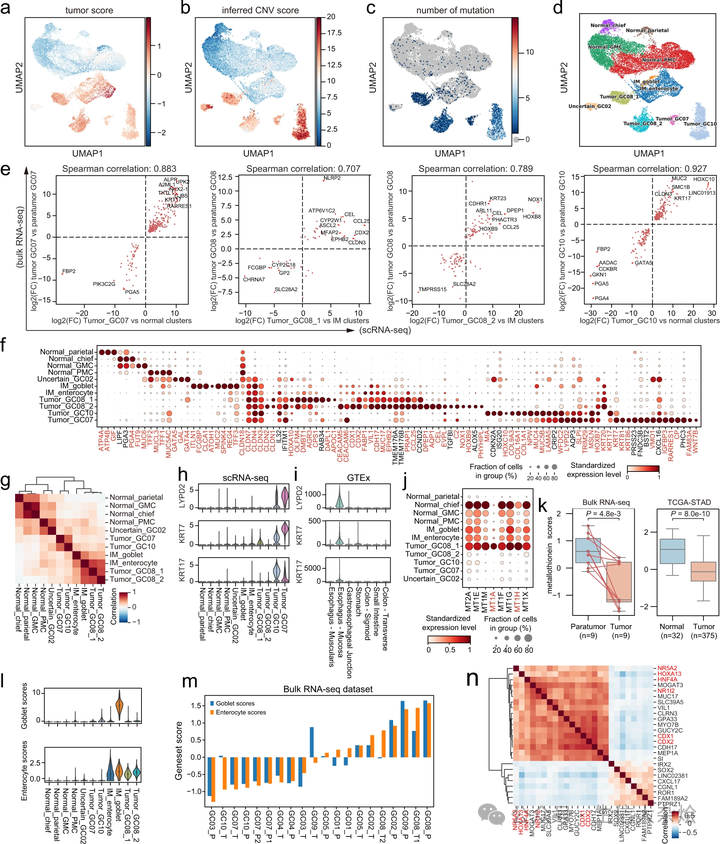
a. 上皮细胞的 UMAP,按肿瘤评分着色;b. 推断的 CNV 评分着色;c. 突变数量着色。d. 上皮细胞的 UMAP。簇以推断的细胞类型标记。GMC 基底腺粘液细胞,PMC 窝粘液细胞,IM 肠化生。e. 散点图显示整体 RNA 测序(肿瘤 vs 肿瘤周围)与 scRNA 测序(肿瘤簇 vs 正常/IM 簇)的 DEG 的 log2 倍变化之间的高相关性。f. 上皮细胞簇的标记基因点图。点的大小表示表达该基因的细胞比例,颜色表示标准化表达水平。红色基因表示在上皮细胞外的细胞检测率低。g. 热图显示上皮细胞簇之间的皮尔逊相关性。小提琴图显示 LYPD2、KRT7 和 KRT17 在 scRNA 测序数据集中的表达(h)和 GTEx 数据集中的胃肠道样本中的表达(i)。j. 点图显示金属硫蛋白相关基因的表达水平。点的大小表示表达该基因的细胞比例,颜色表示标准化表达水平。红色基因表示在上皮细胞外的细胞检测率低。k. 箱形图显示整体 RNA 测序数据集中的金属硫蛋白评分(左;双侧配对 t 检验)和 TCGA-STAD 数据集中的金属硫蛋白评分(右;双侧 Wilcoxon 秩和检验)。所有箱形图:箱子,四分位距(IQR);水平线,中位数;须,最极端值在 ±1.5×IQR 内。l. 小提琴图显示杯状细胞评分(顶部)和肠细胞评分(底部)。m. 条形图显示整体 RNA 测序数据集中的杯状细胞和肠细胞评分。n. 热图显示通过结合我们的 scRNA 测序、整体 RNA 测序和 TCGA-STAD 的整体 RNA 测序生成的相关系数的乘积,展示 CDX2 相关基因的相关性。红色基因被预测为 CDX2 的上游调节因子。
识别促进肠化生的潜在调节因子
为了更好地评估患者的 IM 水平,我们使用细胞类型特征基因(补充表 1)计算了 scRNA 簇和整体数据的杯状细胞评分和肠细胞评分(图 2l,m)。GC08 和 GC09 在肿瘤样本和肿瘤周围样本中都显示出高水平的 IM。尽管 GC07 和 GC10 的肿瘤病理分类分别为肠型和混合型(补充数据 1),但两者的肿瘤都没有表现出明显的肠道基因表达。
CDX2 被认为是 IM 的主要转录因子。为了以一种稳健的方式搜索与 CDX2 相关的基因,我们结合了我们的两个数据集以及癌症基因组图谱(TCGA)的胃腺癌(STAD)数据集,以找到与 CDX2 在表达上相关的基因。从三个数据集中生成的 Spearman 等级相关系数相乘在一起,通过 scRNA-seq 中上皮细胞外的低细胞检测率过滤基因,以避免来自其他细胞类型的混杂效应(见方法)。与 CDX2 高度相关的基因包括已知的 CDX2 靶基因,如 GUCY2C、CDH17、SI 和 GPA33。CDX1 是另一个在远端肠道表达的同源框基因,能够诱导 IM;SOX2 被报道负责胃部特化,并且能够在蛋白水平与 CDX2 相互作用。
除了提供潜在的 CDX2 靶基因外,我们还通过应用 SCENIC 来检查上游调节 CDX2 的转录因子(在图 2n 中以红色显示的),SCENIC 基于基因表达和转录因子基序的富集预测 TF 的下游靶标。HNF4A 先前被报道在 GATA6、TCF4 和 β- 连环蛋白存在时调节 CDX2,而 HOXA13、NR5A2 和 NR1I2 在调节 CDX2 中的功能需要进一步研究。我们在胃癌细胞系中进行了过表达实验,发现 HOXA13 可以在 SGC-7901 细胞中而不是在 MKN-28 细胞中上调 CDX2(补充图 3b,c)。同时,CDX2 的过表达在不同程度上上调了 HNF4A、HOXA13、NR5A2、NR1I2 和 CDX1,并且使细胞形状变长(补充图 2d-f)。根据这些结果,在某些情况下 HOXA13 和 CDX2 可能形成正反馈循环。这个潜在的分子机制值得进一步研究。
GC 中的基质隔间发生了大规模改造
为了解密 GC 中 TME 的基质细胞的功能,我们重新聚类了所有基质细胞,发现肿瘤周围组织和肿瘤组织之间的基质细胞有明显的区别,表明 TME 中的基质细胞与肿瘤周围组织的基质细胞相比经历了全球性的转录组改变(图 3a)。然后,我们将这些基质细胞分为 12 个不同的簇,其中 Endo_1、Fib_1 和 SMC_1 主要富集在肿瘤组织中(图 3b 和补充图 4a)。有趣的是,肿瘤周围的基质细胞比来自肿瘤的基质细胞具有更大的异质性(补充图 4b),暗示基质细胞在正常生理条件下可能具有更多样的功能,但在 TME 中变得专门化。
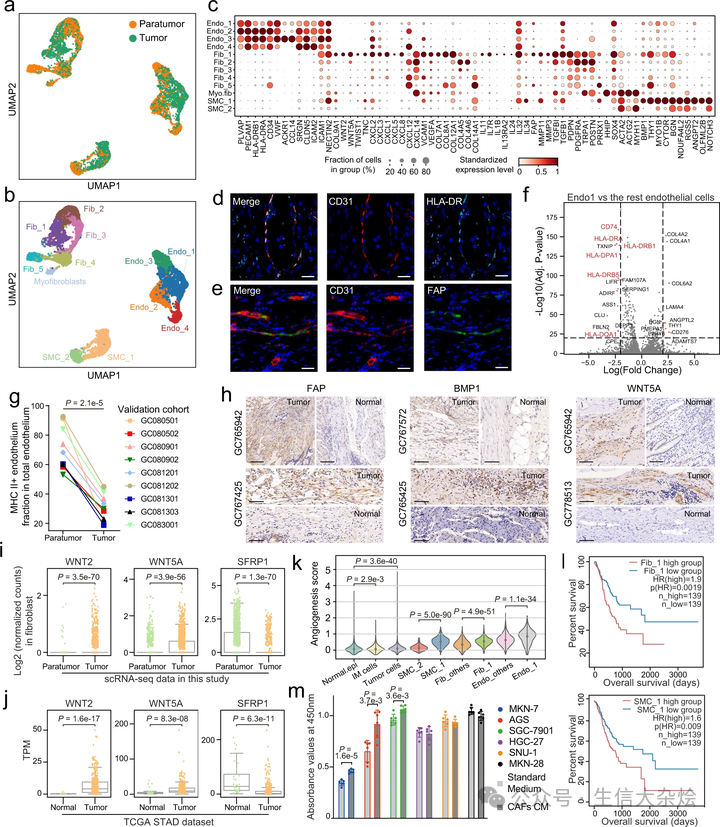
a. 基质细胞的 UMAP,按细胞组织来源着色;b. 推断的细胞类型着色。c. 点图显示基质细胞亚群中的标记基因。点大小表示表达细胞的比例,颜色表示标准化表达水平。d-e. 使用抗 CD31 和抗 HLA-DR 抗体的多色免疫组化(IHC)染色显示 HLA-DR+ 内皮细胞(d;n=6),以及使用抗 CD31 和抗 FAP 抗体显示 FAP+ 内皮细胞(e;n=6)。比例尺代表 20 微米。f. 火山图显示肿瘤富集的 Endo_1 与其他内皮细胞类型的差异表达基因。虚线表示 p 值<1e-20 和|log2(FC)|>2(双侧 Wilcoxon 秩和检验,经 Bonferroni 校正)。g. 点图显示肿瘤周围 MHC II 类 + 内皮细胞比例高于肿瘤内(n=9)(双侧配对 t 检验)。h. 在独立生物标本的福尔马林固定和石蜡包埋切片上进行 FAP、BMP1 和 WNT5A 的 IHC 染色。比例尺代表 100 微米。i. 箱形图显示肿瘤(n=1274)和肿瘤周围(n=1461)成纤维细胞中 WNT 相关基因的表达。每个点代表一个单细胞(双侧 Wilcoxon 秩和检验)。j. 箱形图显示肿瘤(n=375)和正常组织(n=32)中 WNT 相关基因的表达。每个点代表一个样本(双侧 Wilcoxon 秩和检验)。k. 小提琴图显示基质和上皮亚群中的血管生成评分。Normal.epi 和 IM 细胞分别代表正常上皮细胞和肠化生细胞(双侧 Wilcoxon 秩和检验)。l. 通过将患者按照相应细胞类型的高(前 40%)和低(后 40%)比例分层,绘制的总生存期 Kaplan–Meier 曲线。在 TCGA-STAD 队列中,Fib_1 和 SMC_1 的高比例与不良预后相关。HR(风险比)和 p(HR) 是通过 Cox 比例风险模型计算的。m. 收集 GC CAFs 的上清液作为条件培养基(CM),并与六个 GC 细胞系孵育 60 小时。通过 CCK-8 实验确定细胞存活率(n=6)。数据表示为均值 ± 标准差(双侧 t 检验)。
值得注意的是,我们发现胃内皮细胞表达主要组织相容性复合体(MHC)II 类基因,如 HLA-DRA 和 HLA-DRB5(图 3c),这一点通过在 GC 患者肿瘤切片上进行的多色免疫组化(IHC)染色得到了确认(图 3d)。此外,我们发现 Endo_1 的 MHC II 类基因表达下调(图 3f),表明 Endo_1 的内在抗原呈递功能受限。我们进一步对额外九个 GC 患者样本进行了流式细胞术,结果显示肿瘤周围的 MHC II 类 + 内皮细胞比例高于肿瘤内(p<0.001, 学生配对 t 检验)(图 3g 和补充图 4c),与 scRNA-seq 数据中的观察一致。
Endo_1 和 Fib_1 都表达了成纤维细胞活化蛋白(FAP),一种典型的癌相关成纤维细胞(CAF)标记。类似地,我们进行了多色 IHC 染色以验证肿瘤中 FAP+ 成纤维细胞和内皮细胞的存在(图 3e 和补充图 4d)。Fib_1 还表达了其他 CAF 标记,如 MMP3 和 MMP11,以及促进癌症发生的炎症相关成纤维细胞标记(IL11, IL24)(图 3c 和补充图 4g)。Wnt 信号通路中的基因,如 WNT2 和 WNT5A,在肿瘤成纤维细胞中上调,而 Wnt 信号的抑制因子 SFRP1 下调(图 3i)。这些基因在 TCGA-STAD 数据集中也显示了相似的表达模式(图 3j)。注意,这三个基因几乎专门由成纤维细胞表达(补充图 4e)。
此外,Fib_1 细胞表现出 TWIST1-PRRX1-TNC 正反馈途径的上调,已知该途径促进 TME 中 CAF 的激活和扩展。同时,分别促进肿瘤生长和血管生成的骨形态发生蛋白 1(BMP1)和 ANGPT2 在 SMC_1 中表达水平显著更高。IHC 结果验证了 FAP、BMP1、WNT5A 的蛋白表达在肿瘤中上调(图 3h)。此后,我们将这三个肿瘤富集细胞簇 Endo_1、Fib_1 和 SMC_1 定义为肿瘤相关基质细胞(TASCs)。
基因集变异分析发现,在 TASCs 中表达的基因展示出独特的代谢特征,广泛参与与癌症相关的途径(补充图 4f)。值得注意的是,肿瘤进展的关键标志之一的血管生成途径在 TASCs 中显著上调(图 3k)。然后,我们检查了 TASCs 比例与 TCGA-STAD 数据集中患者生存的潜在关联。通过 MuSiC 算法估计了 TCGA-STAD 中的细胞类型比例,该算法利用 scRNA-seq 数据实现整体组织细胞类型解卷积。引人注目的是,我们发现 Fib_1 和 SMC_1 与更差的预后相关(图 3l),并且 Fib_1 和/或 SMC_1 特异性表达的个别基因如 INHBA 和 PLXDC1 也具有潜在的预后能力(补充图 4g, i)。Kumar 等人报道重组 INHBA 足以在正常成纤维细胞系中上调 FAP 和胶原基因的表达。此外,我们的体外实验也显示,来自 CAF 的上清液有能力支持几种胃癌细胞系的肿瘤生长(图 3m)。总之,TASCs 经历了大规模改造,并在 GC 中显示出潜在的促肿瘤特性。
肿瘤中富集了与脂质相关的巨噬细胞
髓系细胞是高度异质的免疫细胞群体,在塑造 TME 中提供了主要贡献。我们在 GC TME 中识别了八个不同的髓系细胞簇,包括两个单核细胞簇、两个巨噬细胞簇和四个树突细胞(DCs)簇(图 4a, b)。我们将两个富集于血液的细胞簇,Mono_CD14 和 Mono_FCGR3A,分别分类为经典的 CD14+CD16- 和非经典的 CD14-CD16+ 单核细胞(补充图 5a, b)。
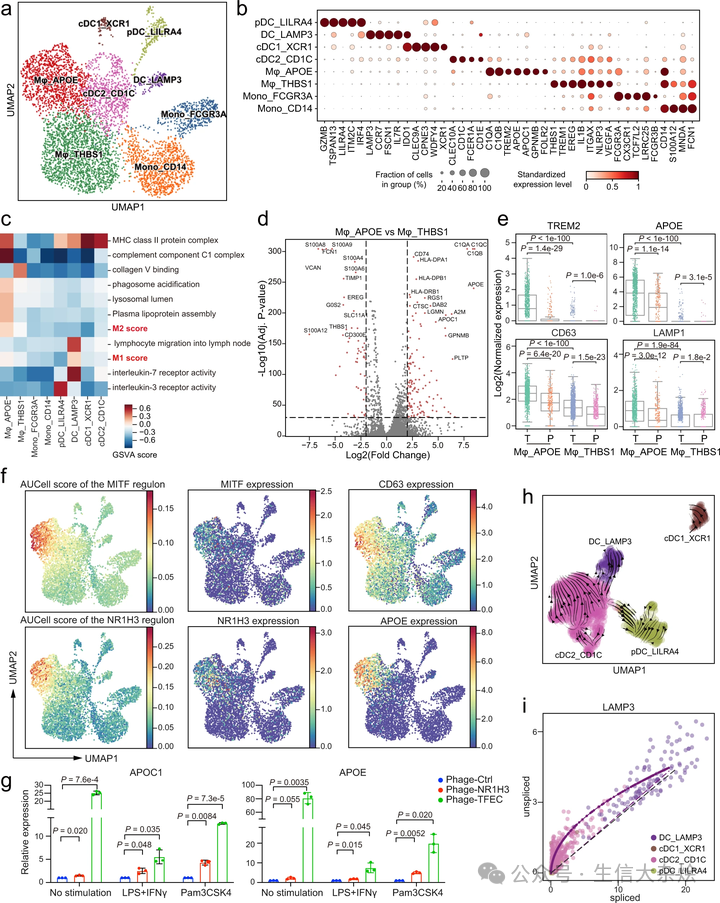
a. 髓系细胞的 UMAP。簇以推断的细胞类型标记。b. 点图显示髓系细胞亚群中的标记基因。点的大小表示表达细胞的比例,颜色表示标准化表达水平。c. 使用 GSVA 评分的不同髓系细胞簇之间的途径活性差异。途径的分数进行了 z 分数标准化。d. 火山图显示 Mφ_APOE 和 Mφ_THBS1 之间的差异表达基因。虚线表示 p 值<1e-20 和|log2(FC)|>2(双侧 Wilcoxon 秩和检验)。e. 箱形图显示巨噬细胞中溶酶体和与脂质相关的基因表达。每个点代表一个单细胞。T 和 P 分别代表肿瘤和肿瘤周围组织。(双侧 Wilcoxon 秩和检验,n=1236 对于 Mφ_THBS1_T,n=926 对于 Mφ_APOE_T,n=544 对于 Mφ_THBS1_P,n=244 对于 Mφ_APOE_P)。f. 髓系细胞的 UMAP,按 NR1H3 和 MITF 的 TF 调控子活性的 AUCell 分数着色,或按基因的标准化表达着色。g. 过表达 NR1H3 或 TFEC 的 THP-1 来源的巨噬细胞被脂多糖(LPS)+ 干扰素 γ(IFNγ)或 Pam3CSK4 刺激。然后通过 qPCR 测量 APOE 和 APOC1 的表达。每列代表三个独立实验的均值 ± 标准差(双侧 t 检验)。h. 通过 RNA 速率推断的 DC 亚群的发育动态 UMAP。i. DCs 中剪接和未剪接 mRNA 的 LAMP3 的速率分析。每个点代表一个细胞。
大多数胃肿瘤 TME 中都存在 Tc17 细胞
为了解析 GC 中 T 细胞的多样性,作者进一步提取并重新聚类了具有 scRNA-seq 数据和配对 TCR 信息的 T 细胞,划分为十个 CD8+ 簇、六个常规 CD4+ 簇(CD4+ Tconv)、三个 CD4+ Treg 簇,以及一个循环簇,代表当前正在细胞周期中进展的 T 细胞(图 5a-c 和补充图 6)。
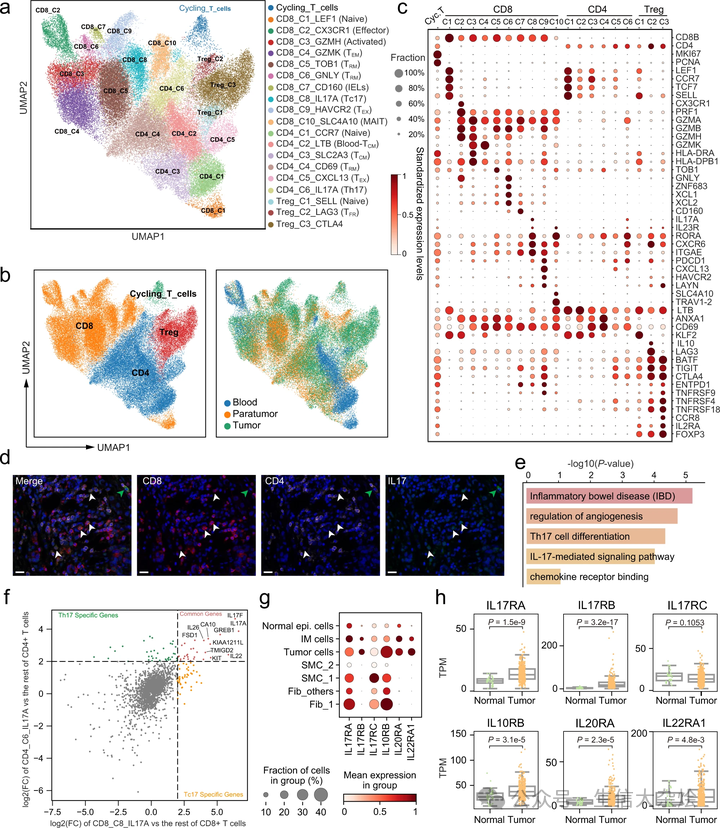
a. 具有 scRNA-seq 数据和配对 TCR 信息的 T 细胞的 UMAP。簇以推断的细胞类型标记。b. 点图显示 T 细胞亚群中的标记基因。点的大小表示表达细胞的比例,颜色表示标准化表达水平。c. T 细胞的 UMAP,左图按细胞类型着色,右图按细胞组织来源着色。d. 使用抗 CD4、抗 CD8 和抗 IL17A 抗体的多色免疫组化(IHC)染色,以患者 GC988401 为例(n=6)。白色和绿色箭头分别指示 CD8+IL17+ 细胞和 CD4+IL17+ 细胞。比例尺代表 20 微米。e. Tc17 中高表达基因富集的 KEGG 术语或途径的条形图,p 值通过超几何分布计算。f. 散点图显示差异表达基因的 log2 倍变化。CD8_C9_HAVCR2 与其他 CD8+ T 细胞(X 轴);Treg_C3_CTLA4 与其他 CD4+ T 细胞(Y 轴)。每个点代表一个基因,内部有颜色注释。g. 点图显示 scRNA-seq 数据集中编码 IL17A、IL17F(IL17RA/IL17RC)、IL22(IL10RB/IL22RA1)和 IL26(IL20RA/IL10RB)的受体基因的表达。h. 箱形图显示 TCGA-STAD 数据集中肿瘤(n=375)和正常组织(n=32)中编码 IL17A、IL17F(IL17RA/IL17RC)、IL22(IL10RB/IL22RA1)和 IL26(IL20RA/IL10RB)的受体基因的表达,按组织来源分组(双侧 Wilcoxon 秩和检验)。
通过 TCR 分析解析 T 细胞亚型的状态转换
接下来,我们试图使用 TCR 克隆信息和拟时序分析来理解各种 T 细胞亚型之间的细胞状态转换。在检测到的 T 细胞中发现了 36,239 个克隆型,其中 30,980 个只被检测到一次(独特 TCR),而 5259 个在两个或更多 T 细胞中被检测到(非独特 TCR)。个别克隆群体的大小范围从 1 到 569(图 6a)。总的来说,CD8+ 簇比 CD4+ 簇有更高程度的克隆扩展,除了原始 CD8+ T 细胞(CD8_C1)(图 6b 和补充图 8a, b)。我们观察到 CD8_C2(效应)、CD8_C3(细胞毒性)、CD8_C4(效应记忆)和 CD8_C10(MAIT)簇既有更高比例的克隆细胞,也有更高比例的克隆细胞在血液和固体组织之间共享 TCR(图 6c)。在克隆性高的簇中,CD8_C2 主要来源于血液,其标记基因富集在与细胞迁移相关的途径(图 6d),因此我们推测 CD8_C2 有从血液渗透到固体组织的潜力。
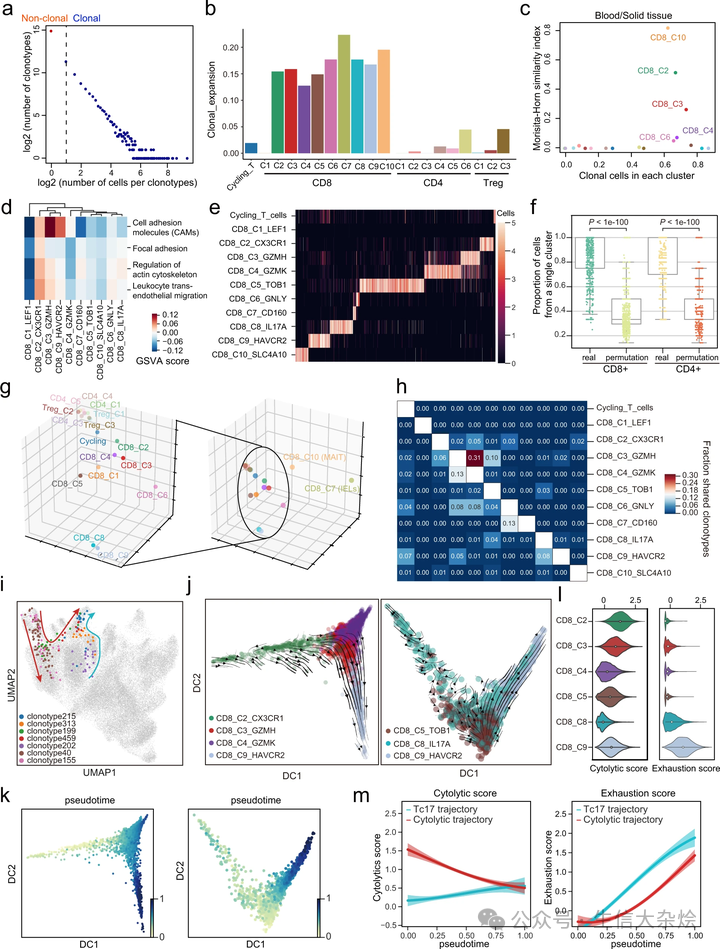
a T 细胞克隆数量与每个克隆型的细胞数量之间的关联。虚线区分了非克隆细胞和克隆细胞,后者通过 TCR 的重复使用被识别。b 条形图显示每个 T 细胞亚群的克隆扩张得分。c 比较每个簇中克隆细胞的比例(x 轴)和通过 Morisita-Horn 相似性指数估计的血液和实体组织之间克隆共享(y 轴)。d 不同 CD8+ T 细胞亚群中与迁移相关的途径活动得分差异,通过 GSVA 评分。e CD8+ T 细胞亚群和周期性 T 细胞中克隆克隆型的分布。颜色越浅表示细胞数量越多;细胞数量上限为 5。f 每个克隆型的单个簇的细胞比例。在簇标签和克隆型标签之间进行了排列(双侧 Wilcoxon 秩和检验,n=1558 对于实际和置换的 CD8+ T 细胞克隆型,n=1046 对于实际和置换的 CD4+ T 细胞克隆型)。g 三维图展示了根据每个簇的 VDJ 基因使用偏差的 T 细胞亚群的 PCA 嵌入。h 热图显示属于主要表型簇(行)的克隆型与其他次级表型簇(列)共享的比例。i 通过选定的 TCR 克隆型对 T 细胞进行着色的 UMAP。红色和青色箭头分别指示血液来源和组织来源 CD8+ T 细胞的状态转换。j 扩散图显示细胞从细胞毒性轨迹(左)和 Tc17 轨迹(右)的 RNA 速度。k 扩散图显示根据 RNA 速度计算的细胞毒性轨迹(左)和 Tc17 轨迹(右)的拟时序。l 小提琴图显示细胞毒性得分(左)和耗竭得分(右)。m 高斯过程回归曲线及其 95% 置信区间显示细胞毒性轨迹(排除 CD8_C4_GZMK)和 Tc17 轨迹的细胞毒性得分(左)和耗竭得分(右)随拟时序的动态。
组织驻留 CD8+ T 细胞通过 Tc17 轨迹达到耗竭状态
为了进一步解密这些簇之间的分化轨迹,我们首先提取在每个潜在轨迹中的簇间共享相同克隆型的细胞,然后使用 RNA 速度分析查询它们在扩散图上的方向性(图 6j,k 和补充图 9a,b)。我们发现从血源 CD8+ T 细胞到耗竭群体存在一个强烈的方向性流动,通过 CD8_C3(细胞毒性)细胞(图 6j,左)。在通向耗竭的轨迹上,T 细胞的细胞毒性得分逐渐降低,耗竭得分逐渐增加(图 6l,m)。与克隆型共享分析结果一致,RNA 速度显示组织驻留 CD8+ T 细胞也通过 Tc17 细胞向耗竭群体展示了一个方向性流动,表明组织驻留 CD8+ T 细胞能够在肿瘤微环境(TME)中分化为 Tc17 细胞,随后产生耗竭表型。
因此,我们将 T 细胞耗竭的两个轨迹命名为“细胞毒性 - 耗竭轨迹”和“Tc17- 耗竭轨迹”。尽管最终达到耗竭状态,我们发现沿 Tc17- 耗竭轨迹的耗竭得分显著高于细胞毒性 - 耗竭轨迹(图 6m)。此外,Tc17 在非幼稚 CD8+ T 细胞中有最低的细胞毒性得分,尽管经历了广泛的克隆扩张,而 CD8_C2(效应)T 细胞在列表中排名第一(图 6b 和补充图 9c)。总的来说,细胞毒性得分沿细胞毒性 - 耗竭轨迹递减,而沿 Tc17- 耗竭轨迹递增(图 6m)。总结来说,我们的观察表明,GC 的 TME 中肿瘤浸润的 CD8+ T 细胞可以通过细胞毒性 T 细胞和 Tc17 细胞达到耗竭状态。
两种耗竭轨迹与不同的转录程序相关联
Tc17 细胞不仅在转录轮廓(图 5a 和补充图 7a)上,而且在 TCR 的 VDJ 基因使用上(图 6g),与细胞毒性 T 细胞(CD8_C3)不同,这表明这两种 T 细胞亚型可能识别两套不同的抗原并在功能上有所不同。我们推测,Tc17 细胞和细胞毒性 T 细胞可能分别产生具有不同转录程序的两种不同类型的耗竭 T 细胞。支持这一观点的是,差异基因分析显示,由 Tc17 细胞衍生的耗竭 T 细胞高度表达角蛋白 KRT86,而由细胞毒性细胞衍生的耗竭 T 细胞高度表达 GZMK(图 7a,b 和补充图 9d)。
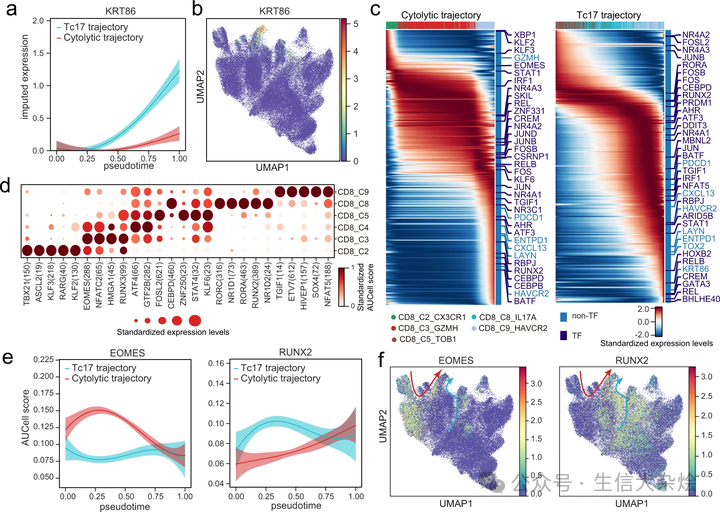
a 使用高斯过程回归曲线和 95% 置信区间展示沿着细胞毒性轨迹(排除 CD8_C4_GZMK)和 Tc17 轨迹的拟时序,KRT86 的动态表达。b 使用 UMAP 展示 T 细胞中 KRT86 的表达。c 热图展示沿细胞毒性轨迹(排除 CD8_C4_GZMK)和 Tc17 轨迹的拟时序,高变异基因的表达。顶部的颜色条代表如图 6g 中的细胞簇;右侧的颜色条注释所有高变异的转录因子(TF)和一些特定的标记基因。d 点图展示通过 SCENIC 计算的 CD8+ T 细胞亚群中 TF 调控元件活性的 AUCell 得分。每个点的大小代表 TFs 的标准化表达水平。e 使用高斯过程回归曲线和 95% 置信区间展示沿着细胞毒性轨迹(排除 CD8_C4_GZMK)和 Tc17 轨迹的拟时序,EMOES(左)和 RUNX2(右)的动态表达。f 使用 UMAP 展示 T 细胞中 EMOES(左)和 RUNX2(右)的表达。红色和青色箭头分别指示血源和组织源 CD8+ T 细胞的状态转换。
肿瘤相关的基质和髓细胞是复杂细胞相互作用的关键媒介
为了解析参与胃癌(GC)的各种细胞类型之间复杂的通信网络,我们接下来通过 CellPhoneDB50 识别了肿瘤和正常组织中的假定细胞 - 细胞相互作用。显然,涉及 TASCs 和巨噬细胞的相互作用在 TME 网络中占主导地位(补充图 10a,b 和补充数据 6)。
聚焦于 TASCs,我们发现肿瘤细胞与基质细胞的相互作用多于正常或类肿瘤上皮细胞(补充图 10c)。Fib_1 为肿瘤细胞表达了丰富的生长因子,如 HGF、FGF7 和 BDNF(图 8a),这与其他研究一致,这些研究显示 HGF-MET 和 FGF7-FGFR4 轴是胃癌及其他肿瘤的有希望的治疗靶标 51,52。值得注意的是,我们发现 HGF 与 TCGA-STAD 数据集中的生存负相关(补充图 10d)。此外,VEGFA 依赖的血管生成和 Ephrin-Eph 双向信号通路也在 Endo_1 和肿瘤细胞中发现。值得注意的是,TASC 亚型展示了紧密的信号网络(图 8b)。例如,由 Endo_1 表达的 TEK 是 SMC_1 表达的 ANGPT2 的受体,表明 SMC_1 参与调节内皮细胞的生存和迁移 53。此外,TASCs 高表达与 Endo_1 和/或 SMC_1 上的 Notch 受体 NOTCH1、NOTCH3 和 NOTCH4 相互作用的 Notch 配体 JAG1 和 DLL1,这与 GO 功能富集分析的结果一致(补充图 10e)。Endo_1 强烈激活了 TNF、VEGF、PDGF、PGF 和 Notch 信号通路(图 8a,b),这些通路广泛参与血管生成的生物过程 54。值得注意的是,TASCs 是这些通路中细胞因子的主要供应者(图 8b)。通过对每种细胞类型进行相关性分析,我们发现在我们的数据集和 TCGA-STAD 队列中,TASC 亚型的比例高度正相关(图 8c 和补充图 11b-d)。
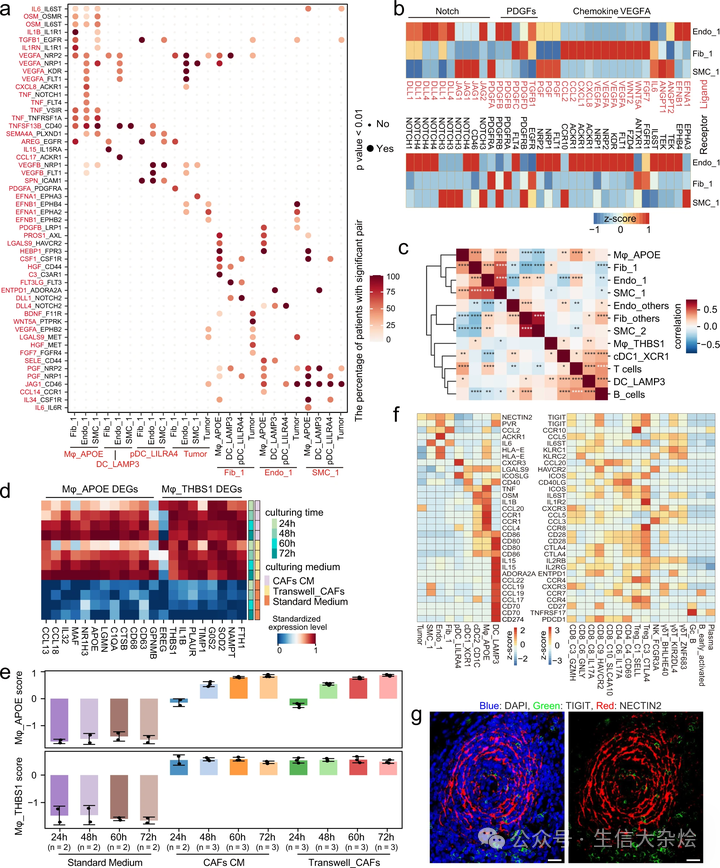
a. 点图展示了肿瘤中选定的配体 - 受体相互作用。细胞亚群在 x 轴上显示;配体(红色)和受体(黑色)对在 y 轴上显示。圆圈的颜色表示在具有这些相互作用细胞亚群的全部患者中,具有显著相互作用(p 值<0.01)的患者比例。p 值由 CellphoneDB 生成,它使用单侧排列测试来计算显著相互作用。b. 热图显示了 Endo_1、Fib_1 和 SMC_1 的选定配体 - 受体对的表达量标准化值。c. 热图显示了 TCGA-STAD 数据集中不同细胞类型推断比例之间的 Spearman 等级相关系数。*P < 0.05, P < 0.01, *P < 0.001, ****P < 0.0001(P 值通过双侧 t 检验计算,确切值可以在源数据中找到)。d. 热图显示了在共培养系统中与胃癌相关成纤维细胞(CAF)共培养的 THP-1 单核细胞衍生的巨噬细胞中,Mφ_APOE 和 Mφ_THBS1 的标记基因的平均表达量,这些细胞分别在胃癌相关成纤维细胞条件培养基(CM)或标准培养基中培养了 24 小时、48 小时、60 小时、72 小时。e. 条形图显示了 Mφ_APOE 和 Mφ_THBS1 的得分。每列代表三次重复的平均值 ±SD。f. 热图显示了肿瘤中淋巴细胞(右侧)和其他亚群(左侧)之间选定的配体 - 受体对的表达量标准化值。g. 使用抗 TIGIT 和抗 NECTIN2 抗体的多色免疫组化染色,以病人 GC769812 为例(n=6)。比例尺代表 20 微米。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









