






2009 英特尔® 线程挑战赛—字符串匹配
问题描述
问题描述:写一个线程程序来搜索用字符串表示的 DNA 序列的数据库,来找到可以匹配的其他 DNA 序列。数据库和查询字符串由四个字符组成:'A', 'C', 'G'. 和 'T' 。对于每个输入的搜索查询字符串,输出必须报告任何输入 DNA 序列内的精确匹配位置。如果查询字符串与数据库内的多个序列匹配,必须报告每个结果;如果查询字符串与同一个数据库序列的多个位置匹配,必须报告完全匹配的那个最早出现的位置。这个问题(资料库档案,查询档案,输出结果)的文件名将在命令行上给出。
文件格式:输入数据库和查询档案将具有相同的格式。每个序列将以大于字符('>')开始,紧跟序列来源的描述,描述不超过 131 个字符。序列会在下一行开始,持续几行。每一行都将包含'A', 'C', 'G'. 和'T'集合的整 80 个字符,除了最后一行,可能会少于80个字符。在最后一行之后将是从下一个序列直至文件末尾的描述符,文件末尾将由描述符(">EOF" )表示 。
对于包含在第二个输入文件的每个查询字符串,输出文件应打印查询序列的描述符和数据库序列的描述符,这个数据库序列包含一个匹配和此匹配在数据库序列内的位置。如果在任何数据库序列内,都没有找到查询序列字符串,应在查询描述符之后打印出一个相关消息来体现这个结果。
计时:总执行时间将用于计分。这允许输入数据库序列的编码或压缩在输入期间完成(如果你的算法采用这种转换)
串行算法
字符串匹配算法分为单模式匹配算法和多模式匹配算法。一个待匹配串为一个待匹配模式,单模式匹配算法一次只进行一个模式的匹配,多模式匹配算法可一次匹配多个模式,常见的单模式字符串匹配算法有Karp-Rabin(KR),Knuth-Morris-Pratt(KMP),Boyer-Moore (BM)等等,多模式算法有Aho Corasick(AC),Wu Manber(WM)等等。
通过一步步了解字符串匹配算法,依次实现了KMP算法,BM算法、WM算法,压缩DNA的WM算法。
其中KMP和BM算法都是利用模式本身包含的信息避免重复对一个字符进行多次比较。其中KMP利用前缀信息加速窗口移动,而BM利用后缀信息加速窗口移动。
WM算法,利用后缀的Hash值对模式进行分组,减少待匹配模式,窗口移动也类似于BM,不同的是它使用Hash值计算Shift表。
由于DNA只包含(A,C,G,T)4个字符,而WM用两个字符计算Hash值,导致冲突过多,效率下降,所以压缩4个字符到1个字节中可减少冲突,提高效率。
压缩后的WM算法仅仅支持长度大于等于8的模式,所以小于8的模式直接用顺序匹配。
并行算法
由于题目是在多个DNA序列中匹配多个模式,所以并行化比较容易,可以使用多个线程分别匹配不同的DNA序列。
单模式匹配算法:
#pragma omp parallel for schedule(guided, 1)
for(i = 0; i < nDatabaseCnt; ++i)
{
XDNASequence& xDNADB = xDatabaseVec.at(i);
for(j = 0; j < nPatternCnt; ++j)
{
XDNASequence& xDNAPattern = xPatternVec.at(j);
pOutput[i * nPatternCnt + j] =
XStrMatch_BM( xDNADB.pSequence, xDNADB.nSequenceLen,
xDNAPattern.pSequence, xDNAPattern.nSequenceLen,
xDNAPattern.pExt);
}
}
根据Amplifier的检测结果进行了优化,将任务粒度减小,提高并行度。
int nCount = nDatabaseCnt * nPatternCnt;
#pragma omp parallel for schedule(guided, 1)
for(i = 0; i < nCount; ++i)
{
const int nIndexD = i / nPatternCnt;
const int nIndexM = i % nPatternCnt;
XDNASequence& xDNADB = xDatabaseVec.at(nIndexD);
XDNASequence& xDNAPattern = xPatternVec.at(nIndexM);
pOutput[i] = XStrMatch_BM( xDNADB.pSequence, xDNADB.nSequenceLen,
xDNAPattern.pSequence, xDNAPattern.nSequenceLen,
xDNAPattern.pExt);
}
多模式匹配算法:
#pragma omp parallel for schedule(guided, 1)
for(i = 0; i < nDatabaseCnt; ++i)
{
XDNASequence& xDNADB = xDatabaseVec.at(i);
XZWMSearch (pXWM, xDNADB.pSequence, xDNADB.nSequenceLen, pOutput + i * nPatternCnt);
}
优化工具
根据测试结果最终选定压缩版本的WM算法提交,以下为ZWM算法的优化结果。
Hotspots检测
使用Intel Amplifier的Hotspots检测功能查找热点函数,结果如下:
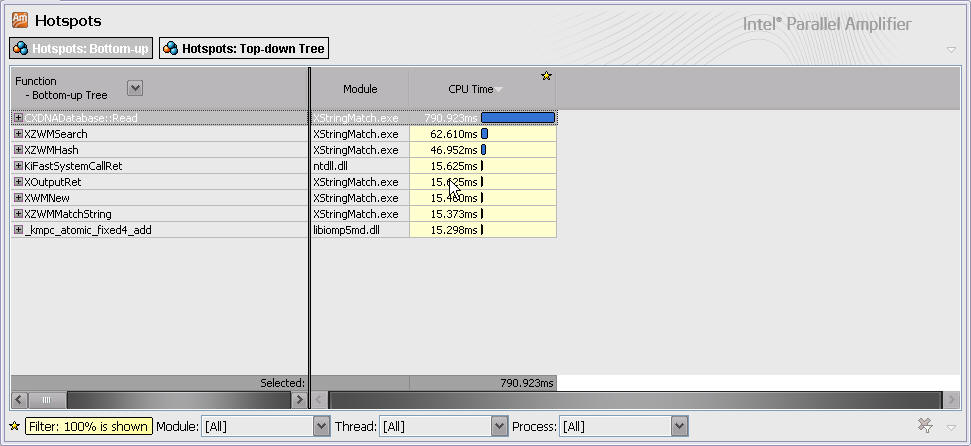
检测结果显示主要的时间开销为读取数据,读取及压缩数据与匹配的时间开销比例为8:1。
Concurrency检测
使用Intel Amplifier的Concurrency检测功能查找可进行并行优化的代码,结果如下:
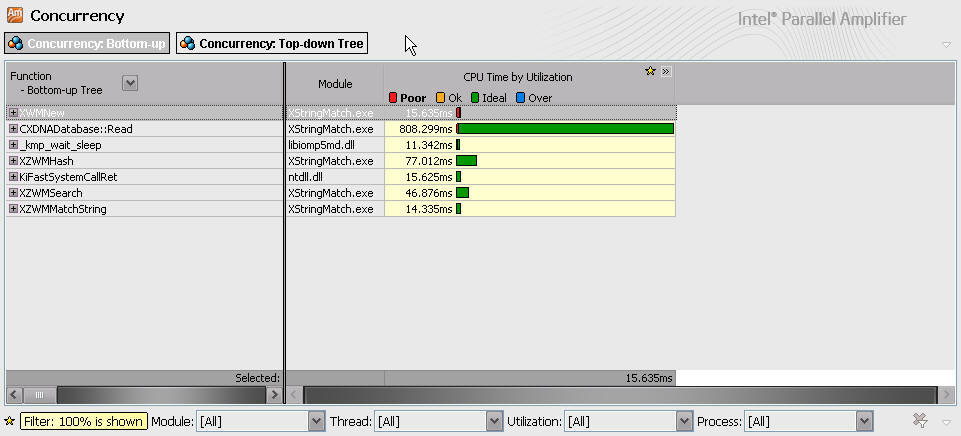
检测结果显示算法的并行度很好, Read函数采用了分块读取数据,每个块的大小必须大于最大DNA序列长度10^6,所以当数据量较小,块数量不够时会导致部分CPU闲置。
Locks and Waits检测
使用Intel Amplifier的Locks and Waits检测功能查找锁和同步等待消耗,结果如下:
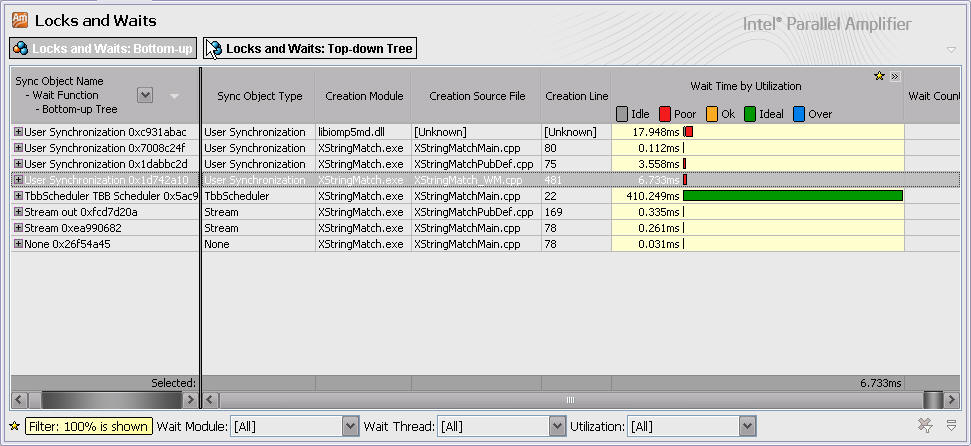
检测结果显示算法几乎不存在同步和锁消耗。
其他优化
1. 使用内存映射结合OpenMp并行的分块加载DNA序列。
2. 使用concurrent_vector保存DNA数据。
3. 使用parallel_sort进行模式的排序。
性能测试
操作系统: 32bit的测试在32位XP下完成。
CPU: Intel(R) Core(TM)2 CPU 5270 @ 1.40GHz
内存: 1G
时间单位: 秒 最短DNA序列长度:32字节
| 测试数据 文件大小 | 测试数据 DNA数量 | 串行 | 并行 | 加速比 |
| 6M_257k | 10_500 | 0.041158 | 0.030744 | 1.34 |
| 49.5M_257k | 100_500 | 0.330507 | 0.213908 | 1.54 |
| 49.5M_2.5M | 100_5000 | 0.565867 | 0.363544 | 1.56 |
| 250M_2.5M | 500_5000 | 2.814094 | 1.711416 | 1.64 |
编译说明
Windows平台:
使用VS2008和Intel Parallel Studio
1. 用VS2008打开本项目.
2. 选择Win32平台Release编译.
3. 进入Bin目录执行文件为XStringMatch.exe.
Linux平台:
使用ICC和TBB
1. 上传压缩包种的Src和Linux两个目录到服务器上.
2. 进入XStringMatch/Linux目录 执行make
3. 进入XStringMatch/Bin目录 执行文件为XStringMatch.
其他:
主办方请使用Win32平台Release版本测试,谢谢!
优化结论
通过解决本题进一步了解了串匹配算法,串匹配算法可以说有数十种之多,但多数算法的基本原理都是利用已经比较出的结果,结合模式本身的信息,尽可能的滑动比较窗口。
TBB提供的concurrent_vector是一个高性能的、线程安全的并行vector类,在并行读取DNA序列时,各个线程需要把读完的DAN序列压入vector,存在并发访问,如果使用std::vector必须做同步,而concurrent_vector则不用同步,非常方便。
致谢
感谢Clay Breshears在论坛上所做的解答,感谢Mu,Pryce为本文章发表到ISN所做的工作,感谢Xia, JeffX P为此解决方案进行的认真细致的翻译。















 1229
1229

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









