







前言
本文主要记录一些pymol使用中的技巧,文章内容不定时更新
显示结构可信度
在Uniprot下载的AlphaFold预测结构或自己从Colab得到的预测结构中,一般都会显示结构的可信度,如下图中的p53蛋白在Uniprot中的预测结构:

但直接下载PDB文件并在pymol中打开是不会显示pLDDT分数的,只需使用下面这一命令即可显示:
spectrum b, red_yellow_green_cyan_blue, minimum=50, maximum=90
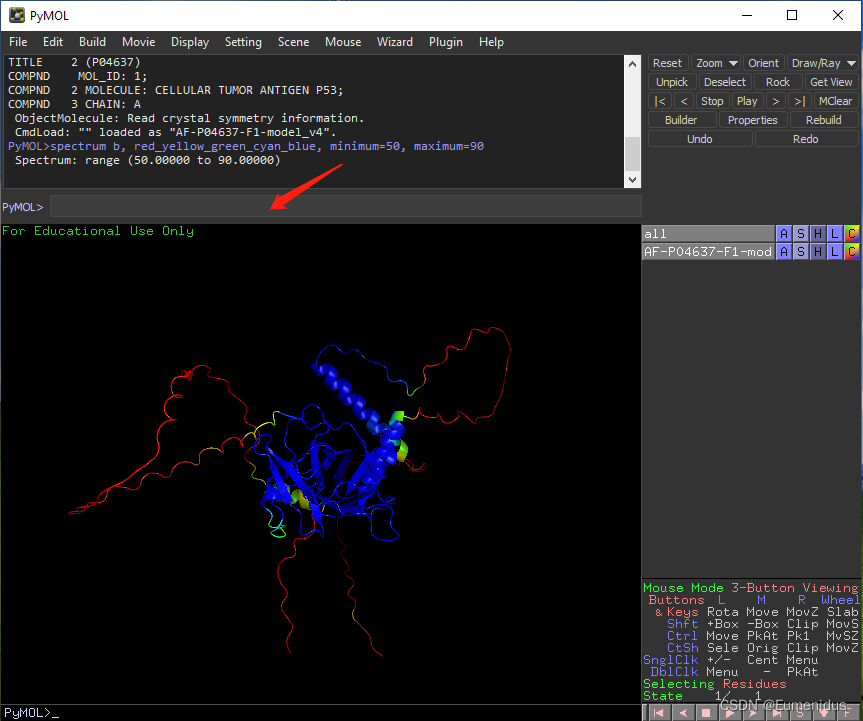
箭头所指向之处即为输入命令的窗口。
通过调整上述命令中的颜色单词还可以实现不同分数对应不同颜色。
输出高清图片
输入如下命令即可得到高清图片:
png filename.png 4000,4000,ray=1
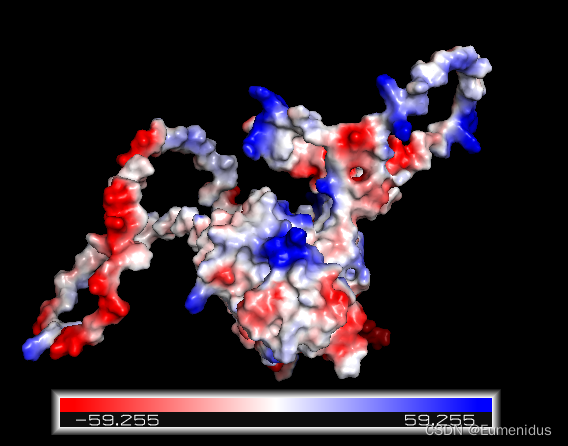
显示表面静电势
依次点击如下按钮即可:
Action -> generate -> vaccum electrostatics -> Protein contact protential (local)
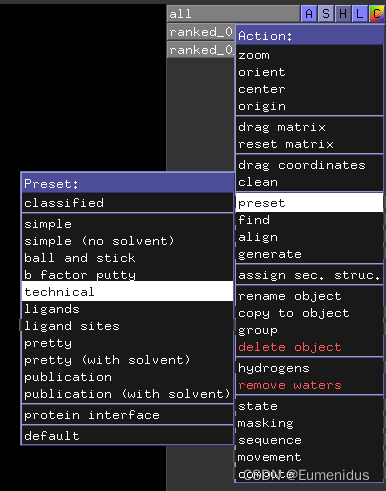
显示氢键
依次点击如下按钮即可:
Action -> preset ->technical
此时蛋白会显示很多黄色小短线,表示的就是氢键的供体和受体
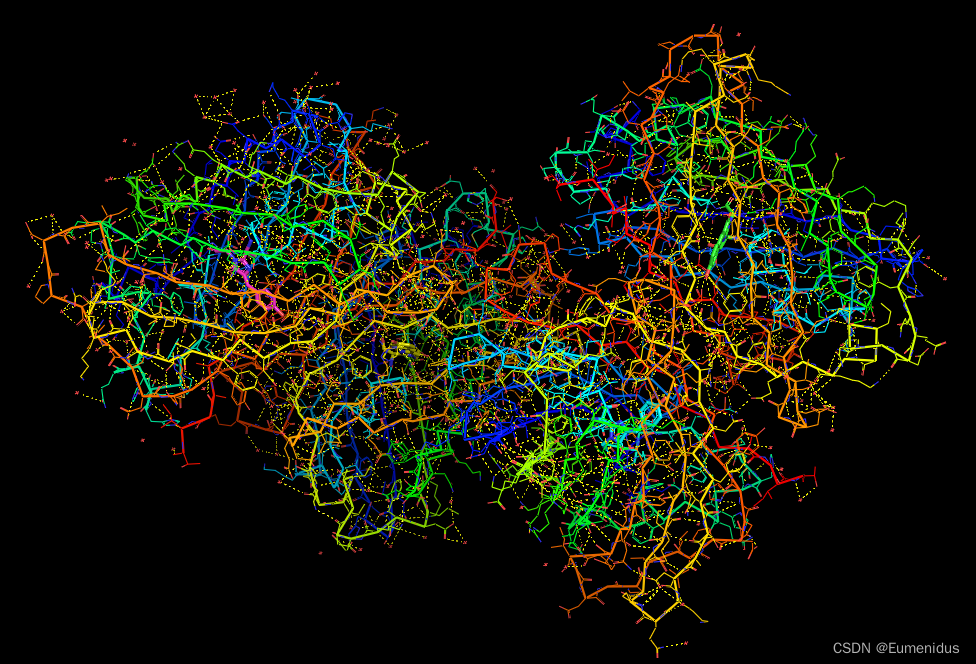
此时的展示形式略显混乱,可以根据蛋白链来标注颜色并用stick来显示模型
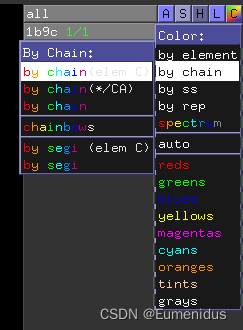
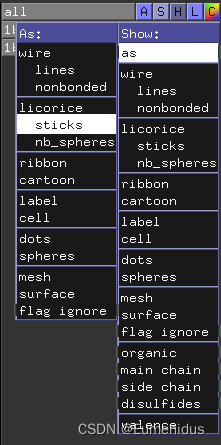
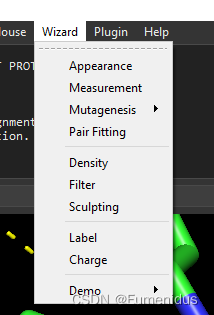
同时使用wizard->measurement来测量两个原子之间的距离,一般来讲氢键要求的距离要短于3.5埃
输出二级结构到文件
在pymol命令行中输入如下命令
open(“ss.txt”,“w”).writelines( [“Residue %s: %s\n”%(a.resi,a.ss) for a in cmd.get_model(“protein_name”+" and n. ca").atom] )
将上面的protein_name替换成自己蛋白的名称即可
运行之后,在该pdb文件的同一路径下就会有ss.txt文件记录着二级结构信息
批量输出二级结构
如果有多个pdb文件需要输出二级结构,可以利用pymol来运行python脚本来获取
将所有需要提取二级结构的pdb文件放到同一个目录下,并新建一个.py文件,命名为script.py(程序名可随意修改,不带空格即可),如下图

在script.py中写入如下程序:
import pymol
from pymol import *
import os
with open("secondary_structure.txt",'wt') as outfile:
for filename in os.listdir():
if filename.endswith(".pdb"):
protein_name=filename.replace(".pdb",'')
cmd.load(filename)
outfile.write(f"{protein_name}\t")
for a in cmd.get_model(protein_name+" and n. ca").atom:
outfile.write(f"{a.ss}")
outfile.write("\n")
cmd.delete(protein_name)
使用pymol打开其中一个pdb文件并在命令行输入
run script.py
回车并运行,即可在同一目录下找到带二级结构的txt
随缘更新。。。。















 1万+
1万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









