







下载
通过NCBI FTP下载本地blast软件:
https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
对于Windows用户,直接下载exe可执行文件即可:
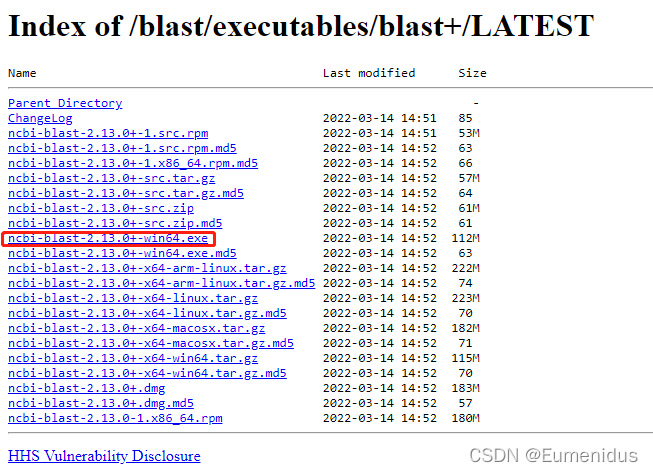
然后按照正常软件安装程序安装即可,可自定义安装路径
提示:以下是本篇文章正文内容,下面案例可供参考
检查blast是否添加至环境路径
一般来说新版的blast都会自动添加到环境目录中,这一步一般不用检查,只是为了以防万一,毕竟有些老版本的blast是不能自动添加到环境目录中的,需要手动添加一下
键盘同时按WIN+R,输入sysdm.cpl,
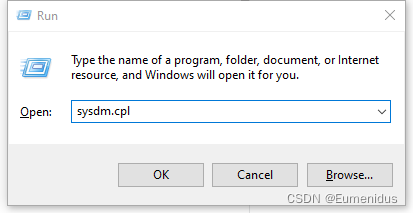
(用的不是自己的电脑,忽略我的英文操作系统)
然后按回车,跳转,选择上方第三个选项卡高级,点下方的环境变量:
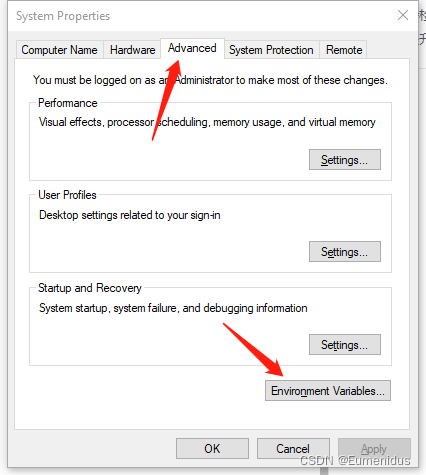
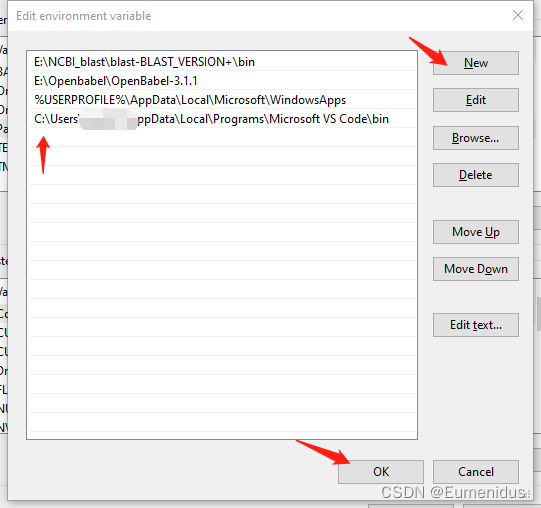
检查你安装blast目录下的bin文件夹是否有在用户PATH中:
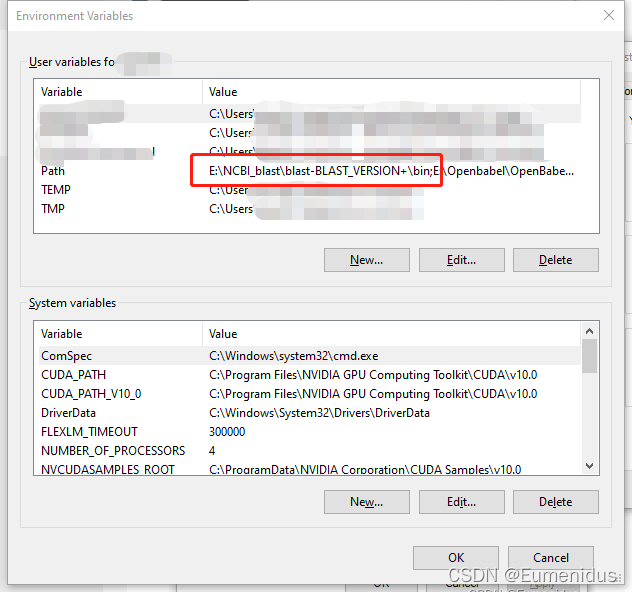
若环境变量中没有blast目录,则需要手动添加一下:
选中PATH一栏,点击Edit,在弹出窗口中选择NEW,然后在下方栏目中复制粘贴你bin文件的路径,再点OK即可添加
使用blast
首先通过NCBI或Uniprot等数据库下载到自己需要的基因组/蛋白组文件,你的目标序列会在这些基因组/蛋白组文件中进行查询
合并fasta文件
下载完成后,如果你有多个物种的基因组\蛋白组需要检索,则需要将这些物种的基因组fasta文件合并为一个,这里使用了一个简单的python脚本来合并:
使用方法,将需要合并的所有文件放在一个文件夹中,文件夹中不要放置其他文件,同时在文件夹中放入这个python脚本,运行即可。
import os
outname="database.fasta"
with open(outname,'wt') as outfile:
for filename in os.listdir():
dot=filename.find(".")
if (filename[dot+1:]=='txt' or filename[dot+1:]=='fasta') and filename!=outname:
species=filename[:dot].replace(" ","_")
with open(filename,'rt') as infile:
for line in infile:
if line[0]=='>':
line=line.split('|')
outfile.write(">{}_{}\n".format(line[1],species))
else:
outfile.write(line)
print("Finish {}".format(filename))
print("Finish all!")
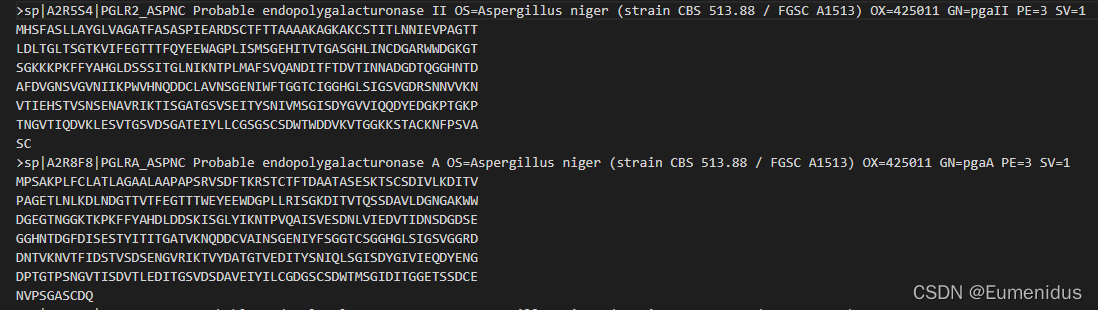
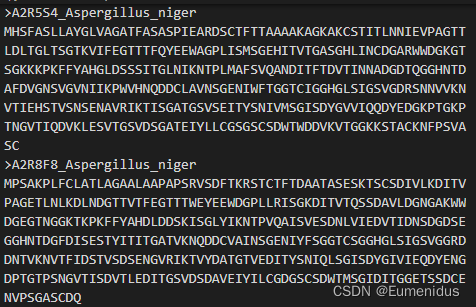
以Uniprot下载得到的蛋白组fasta为例,其序列名都很长,因此我在合并文件时还加入了截短序列名的功能,这里是直接截取了序列名中两个|中间的序列号,同时加入fasta的文件名,因此建议将蛋白组文件名直接命名为其物种名,且尽量只有属名和种加词两个单词
修改前:
修改后:
检查重名
注意:检索的基因组中不能有重复的序列名,否则在建库的过程中会报错
这里同样使用了一个简单的python脚本来检查是否有重复的序列名:
with open("database.fasta",'rt') as infile:
namelist=[]
repeat=False
for line in infile:
if line[0]=='>':
line=line.strip("")[1:]
i=0
while i<len(namelist):
if namelist[i]==line:
repeat=True
i=-1
print("序列{}有重复".format(line))
break
elif namelist[i]<line:
break
else:
i+=1
if i>0:
namelist.insert(i,line)
if not repeat:
print("该文件中没有重复的序列")
blast建库
此处建议新建一个空文件夹,将你合并好的蛋白组fasta和要查询的序列fasta放到这个文件夹中,在地址栏输入cmd,回车
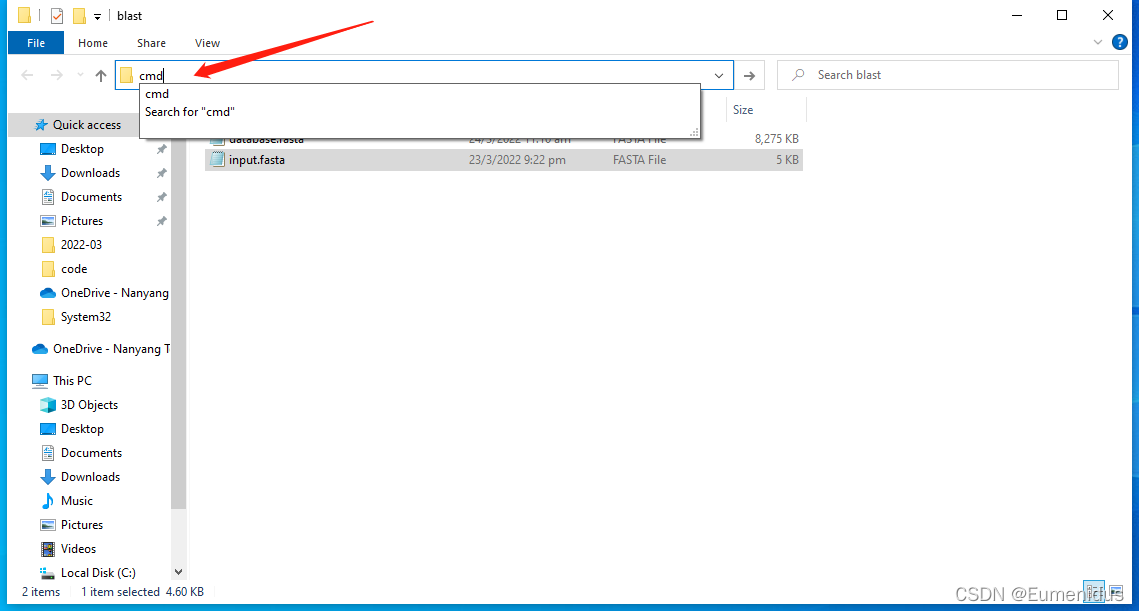
在命令行界面中,输入如下命令进行建库
makeblastdb.exe -in database.fasta -parse_seqids -hash_index -dbtype prot
其中database.fasta需要更换为你自己的蛋白组文件名
若你构建的是基因组文件,则需要更换数据类型为核酸:
makeblastdb.exe -in database.fasta -parse_seqids -hash_index -dbtype nucl
建库完成后,会出现一大堆文件:
blast检索目标序列
如果你需要检索的fasta序列已经放置在数据库同一目录下,则可以在上面的cmd中继续输入:
blastp.exe -task blastp -query input.fasta -db database.fasta -out blast_result.txt -outfmt 6 -evalue 1e-5
如果需要比对的是核酸,则使用blastn
blastn.exe -task blastn -query input.fasta -db database.fasta -out blast_result.txt -outfmt 6 -evalue 1e-5
参数说明:
-query 需要检索的序列文件名
-db 需要在哪个数据库中检索
-out 输出文件名
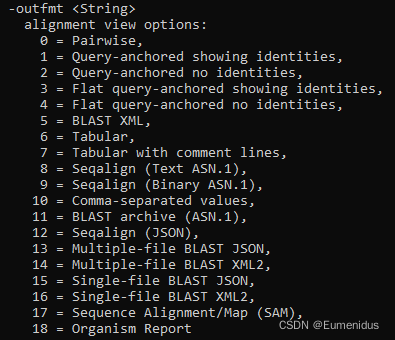
-outfmt 输出格式,一般选6即可,有0-18可选
-evalue E值阈值,高于这个E值的序列不输出到结果中,默认值为10,建议设到10-5以下
除此之外,还可以加入num_thread等参数,表示同时使用多少个线程来计算。其他更多参数可以使用
-help来查看:
blastp.exe -help
可选输出格式:
输出结果
如果选择的输出格式是6,那么得到的结果会是制表符分隔的表格,使用excel打开即可:
得到的一般会是这样一个有13列的表格
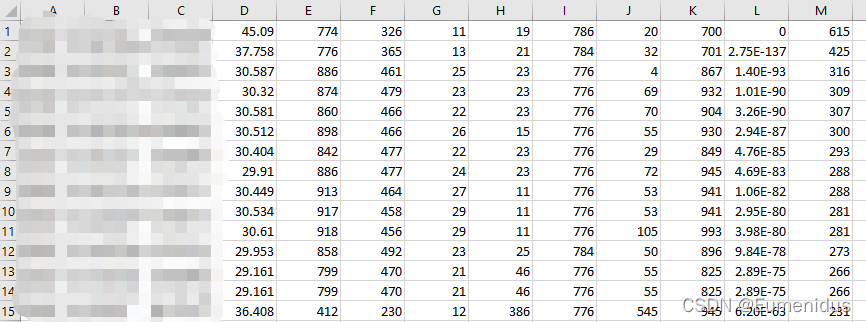
这几列对应的表头是:
query id:查询序列ID标识
subject id:比对上的目标序列ID标识
identity:序列比对的一致性百分比
alignment length:符合比对的比对区域的长度
mismatches:比对区域的错配数
gap openings:比对区域的gap数目
query start:比对区域在查询序列(Query id)上的起始位点
query end:比对区域在查询序列(Query id)上的终止位点
subject start:比对区域在目标序列(Subject id)上的起始位点
subject end:比对区域在目标序列(Subject id)上的终止位点
e-value:表明在随机的情况下,其它序列与目标序列相似度要大于S值的可能性,e-value的分值越低越好
bit score:比对结果的bit score值
后续就可以根据这些blast结果从中找到自己感兴趣的序列来进行分析
提取序列
由于需要根据序列号从基因组/蛋白组文件中逐个找回序列的工作十分繁琐,这里同样使用一个简单的python脚本来进行批量查找:
import os
print("请在本文件夹下input.txt文件中输入需要查找的序列,按回车分隔")
print("待查找的序列需与该程序置于同一文件夹中,且后缀名只能为.txt或.fasta")
try:
with open('input.txt', 'rt', encoding='utf-8') as input_sequence_file:
search_name=[n.strip() for n in input_sequence_file.read().strip().split("\n")]
except IOError:
print("本文件夹下没有input.txt文件")
outfilename="result.fasta"
with open(outfilename,'wt') as outfile:
for search_file_name in os.listdir():
filetype=search_file_name[search_file_name.find(".")+1:]
if (filetype!='txt' and filetype!='fasta') or search_file_name=='input.txt' or search_file_name==outfilename:
continue
with open(search_file_name,'rt') as search_file:
key=''
for line in search_file:
if line[0]=='>':
if len(key)>0:
outfile.write(">{}\n{}\n".format(key,seqdict[key]))
key=''
if line[1:].strip() in search_name:
key=line[1:].strip()
seqdict={key:''}
elif len(line.strip())>0 and len(key)>0:
seqdict[key]+=line.strip()
使用方法:将要查找的序列名放在input.txt中,每行一个;然后将数据库fasta文件和input.txt放在同一个文件夹目录下,不要放置其他多余文件,将该脚本放置在该文件夹中,运行,得到的结果会输出在output.txt中

















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









