






文章链接:https://www.nature.com/articles/s41467-021-27137-3
代码:A unified DTI prediction framework based on knowledge graph and recommendation system | Zenodo## A unified DTI prediction framework based on knowledge graph and recommendation system # Code and data description ## Scripts - `kge_nfm.py`: the complement of the KGE_NFM & NFM methods. - `kge_rf.py`: the complement of the KGE_RF & RF methods. - `deepdit.py`: the complement of the MPNN_CNN & DeepDTI methods. - the complement of DTINet and DTiGEMS is tested based on their source packages (more in Prerequisites) ## `data/` directory #### `yamanishi_08/` directory - `data_folds/`: 10 folds training set and test set in the three scenarios - `warm_start_1_1/` - `warm_start_1_10/` - `drug_coldstart/` - `protein_coldstart/` - `kg_data/`: supporting knowledge graph data - `dt_all_08.csv`: whole DTI dataset - `791drug_struc.csv`: drugbank id and smiles of drugs - `989proseq.csv`: kegg id and sequences of proteins - `morganfp.txt`: list of drug morgan fingerprints - `pro_ctd.txt`: list of protein descriptors #### `BioKG/` directory - `data_folds/`: 10 folds training set and test set in the three scenarios - `warm_start_1_10/` - `drug_coldstart/` - `protein_coldstart/` - `kg.csv`: supporting knowledge graph data - `dti.csv`: whole DTI dataset - `comp_struc.csv`: drugbank id and smiles of drugs - `pro_seq.csv`: sequences of proteins - `fp_df.csv`: list of drug morgan fingerprints - `prodes_df.csv`: list of protein descriptors #### `hetionet/` directory - `data_folds/`: 10 folds training set and test set in the three scenarios - `warm_start_1_10/` - `drug_coldstart/` - `protein_coldstart/` - `kg.csv`: supporting knowledge graph data - `dti.csv`: whole DTI dataset - `map_drugs_df`: drugbank id and smiles of drugs - `pro_seq.csv`: sequences of proteins - `fp_df.csv`: list of drug morgan fingerprints - `prodes_df.csv`: list of protein descriptors #### `luo's_dataset/` directory - `data_folds/`: 10 folds training set and test set in the three scenarios - `warm_start_1_1/` - `warm_start_1_10/` - `drug_coldstart/` - `protein_coldstart/` - `mapping/`: related mappings and similarity matrix (https://github.com/luoyunan/DTINet) - `protein.txt`: list of protein names - `disease.txt`: list of disease names - `se.txt`: list of side effect names - `drug_dict_map`: a complete ID mapping between drug names and DrugBank ID - `protein_dict_map`: a complete ID mapping between protein names and UniProt ID - `Similarity_Matrix_Drugs.txt` : Drug similarity scores based on chemical structures of drugs - `Similarity_Matrix_Proteins.txt` : Protein similarity scores based on primary sequences of proteins - `feature/`: related features used in methods - `drug_smiles.csv`: drugbank id and smiles - `seq.txt`: list of protein sequences - `morganfp.txt`: list of drug morgan fingerprints - `pro_ctd.txt`: list of protein descriptors #### `eg_model/` directory We provided a pre-trained kge model for example. - `dismult_400_warm_1_10.pkl` # Prerequisites #### Operating system: Linux #### Programing language: python #### KGE_NFM & NFM dependencies ``` - python 3.6 - pandas '1.1.5' - numpy '1.18.4' - scikit-learn '0.24.1' - tensorflow '1.15.0' - ampligraph '1.3.2' - deepctr '0.8.4' ``` #### baseline dependencies - RF & KGE_RF (included in KGE_NFM&NFM dependencies) - MPNN_CNN & DeepDTI: - source: https://github.com/kexinhuang12345/DeepPurpose ``` - deeppurpose '0.0.9' - torch '1.6.0+cu101' ``` - DTINet: - source: https://github.com/luoyunan/DTINet - note: in this work, we run the DTINet in a python environment, which need Linux system and python2. Importantly, this method requires the [Inductive Matrix Completion](http://bigdata.ices.utexas.edu/software/inductive-matrix-completion/) (IMC) library. More detailed information about the installation of this method could be found in the source code of the DTINet. - DTiGEMS: - source: https://github.com/MahaThafar/DTiGEMSplus - TriModel: - source: http://drugtargets.insight-centre.org/ # Example (kge_nfm.py) #### A brief presentation of the results: - return average loss when training kge model ``` Average Loss: 0.475181: 2%|###3 | 1/50 [01:10<57:31, 70.44s/epoch] ``` - return performance(mrr) on training set of DTI for early stopping (kge_model in `eg_model/`) ``` In [35]: roc = roc_auc(test_label,test_score) ...: pr = pr_auc(test_label,test_score) ...: print(roc) ...: print(pr) 0.8731770833333332 0.44079654835037246 ``` - nfm training process (`patience=10`) ``` In [45]: roc_nfm,pr_nfm,pred_y = train_nfm(feature_columns,train_model_input,train_label,test_model_input,test_label,patience) Train on 44851 samples Epoch 1/2000 44851/44851 - 2s - loss: 0.5332 - precision: 0.0976 Epoch 2/2000 44851/44851 - 1s - loss: 0.4143 - precision: 0.0000e+00 Epoch 3/2000 44851/44851 - 1s - loss: 0.3456 - precision: 0.0000e+00 Epoch 4/2000 44851/44851 - 1s - loss: 0.3443 - precision: 0.0000e+00 Epoch 5/2000 44851/44851 - 1s - loss: 0.3470 - precision: 0.0000e+00 Epoch 6/2000 44851/44851 - 1s - loss: 0.3382 - precision: 0.0000e+00 ...... Epoch 279/2000 44851/44851 - 1s - loss: 0.0758 - precision: 0.9248 Epoch 280/2000 44851/44851 - 1s - loss: 0.0753 - precision: 0.9327 Epoch 281/2000 44851/44851 - 1s - loss: 0.0796 - precision: 0.9155 Epoch 282/2000 44851/44851 - 1s - loss: 0.0764 - precision: 0.9276 Epoch 283/2000 44851/44851 - 1s - loss: 0.0739 - precision: 0.9127 ``` - reutrn results as type of roc_auc & pr_auc ``` 0.9812476679104477 0.8803416284646345 ```https://zenodo.org/record/5500305#.Y6FohOhByUkA unified DTI prediction framework based on knowledge graph and recommendation system | Zenodo
研究背景:
药物目标相互作用(DTI)的预测在各个领域的药物开发中起着至关重要的作用,例如虚拟筛查,药物重新利用和鉴定潜在的药物副作用。基于生物实验的传统药物靶标验证方法精度低、成本高、周期长,因此通过计算模拟的方法预测药物和靶标间的相互作用是非常必要的。在这里,我们开发了KGE_NFM,这是通过组合知识图(KG)和推荐系统的统一预测框架。该框架首先学习了KG中各个实体的低维表示,然后通过神经分解机(NFM)整合了多模式信息。KGE_NFM在三个现实情况下进行评估,并在四个基准数据集上实现准确,可靠的预测,尤其是在蛋白质冷启动的情况下。
药物-靶标相互作用预测的方法大致分为三类:
(1)基于结构的方法,使用药物和靶点蛋白的3D结构来计算模拟两者的结合情况,这种方法受限于可利用的靶点蛋白结构;
(2)基于配体的方法,其基于化学相似性原理,即“相似的化学分子具有相似的生物学效应”,但由于蛋白信息未用于预测,当目标蛋白的已知相关配体数量较少时,这类方法的预测性能会大大降低;
(3)化学基因组学方法,它可以同时利用药物和蛋白质的信息来进行相互作用预测,有望克服前两类方法的缺点,但由于药物相互作用数据集不可避免的高稀疏性,其在不同应用场景下的预测精度仍有待提高。
本研究提出的KGE_NFM方法通过将知识图谱和推荐系统相结合,构建了一个系统化的DTI预测框架。此框架首先通过知识图谱表征方法学习图谱中各种药物相关概念实体的低维表示,然后通过神经因子分解机集成知识图谱中所学到的多组学信息和药物与蛋白的结构表征信息实现DTI的预测。
2.KGE_NFM方法
模型框架
KGE_NFM 可以分为两个部分:(1)知识图谱的构建与表征向量的提取;(2) 基于神经因子分解机的多维度特征信息整合。KGE_NFM 的基本框架如图1所示。
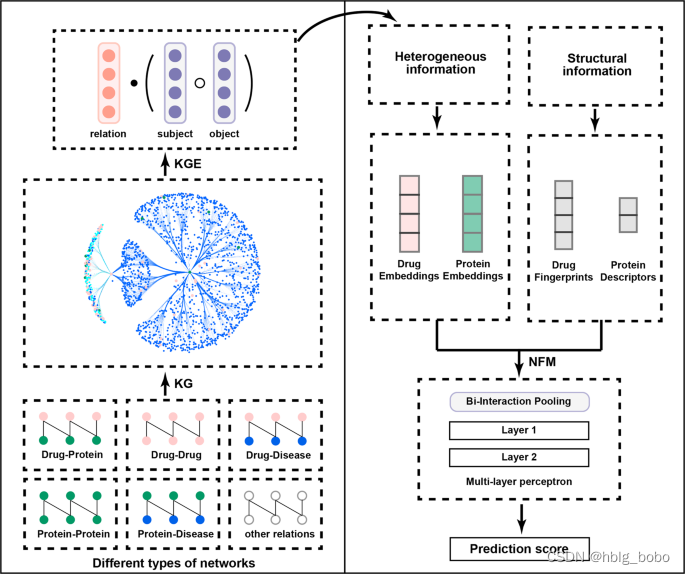
KGE_NFM 的输入包括药物-靶标相互作用数据以及来源于不同组学的相关数据,其储存及输入格式为三元组。每个三元组包括头实体、关系及尾实体,头实体和尾实体相当于知识图谱中的节点,而关系则相当于知识图谱中的边。得到整合的知识图谱后,通过DisMult表征方法将知识图谱中的实体和关系以稠密向量的方式提取出。然后,将知识图谱中提取得到的表征向量作为异构信息的输入,将药物的分子指纹和蛋白的描述符作为结构信息的输入,通过embedding层和Bi-Interaction层提取交互信息,最后将这三类信息整合,通过全连接神经网络来获得输出。
数据集
在这项研究中,四个基准数据集(Luo’s dataset, Hetionet, Yamanishi_08’s dataset and BioKG)被用于比较KGE_NFM与其他最先进的DTI预测方法。Luo’s dataset由四种类型的节点(即药物、蛋白质、疾病和副作用)和六种类型的边(即药物靶点相互作用、药物-药物相互作用、蛋白质-蛋白质相互作用、药物-疾病关联、蛋白质-疾病关联和药物副作用关联)组成。网络总共包含 12015 个节点和1895445条边。
Hetionet整合了29个不同来源的生物医学数据库,网络总共包含11种不同类型的47031个节点(小分子化合物,蛋白,生物过程、分子功能、细胞成分等)和24种不同类型的2250197条边。
Yamanishi_08的DTI数据集由四个子数据集组成:酶(E)、离子通道(IC)、G-蛋白耦合受体(GPCR)和核受体(NR)。知识图谱中的辅助信息包括ATC,BRITE,疾病和pathway等。网络总共包含25487 个节点和95579条边。
BioKG是一个专为关系学习而设计的生物知识图谱,集成了来自 14 个数据库的生物医学数据。知识图谱中包含105524个的节点和2043846条边。
实验任务
为了评价KGE_NFM,作者设计了三种验证场景来测试更贴近真实情况下的预测性能,分别为热启动(warm start),对于药物的冷启动(cold start for drugs),对于蛋白的冷启动(cold start for proteins)。在三个测试场景下,作者将数据集,即活性数据(正样本)按照 1:10 的比例随机划分为测试集和训练集。完成训练集和测试集的划分后,再分别为训练集和测试集构建了负样本,使训练集和测试集中正负样本比例约为1:10。对于训练集与测试集的划分以及负样本的构建,具体的流程可参考图2。在热启动测试场景下,作者在划分数据时保证测试集中的药物靶标相互作用对所涵盖的药物和靶标蛋白在训练集中都出现过;对于药物的冷启动,保证测试集所涵盖的药物在训练集中都不存在;对于蛋白的冷启动,保证测试集所涵盖的靶标蛋白在训练集中都不存在。负样本的构建流程对于三个场景都是相同的,具体的流程为:对于数据集中存在的每个药物,分别与整个基准数据集中存在的所有靶标蛋白组成关系对,然后随机从中选取10个关系对,在所有构造的关系对中过滤掉可能会出现的正样本,去除重复项。

3.实验结果
作者将KGE_NFM与三种不同类型的 DTI 预测方法(即基于特征的方法、端到端的方法和异构数据驱动的方法)进行了比较,如表一所示。在结果分析中,作者重点关注了Yamanishi_08和 BioKG 数据集的性能评估,以比较具有不同数据量但类似数据结构的知识图谱对DTI预测结果的影响。

Yamanishi_08’s dataset结果评价
在热启动的情况下,作者发现异构数据驱动方法DTiGEMS+、Trimodel和KGE_NFM在正负样本平衡和不平衡的两种情况下都有稳健的预测性能。当数据集的正负样本平衡时,基于特征的方法,包括RF(AUPR=0.901)和NFM(AUPR=0.922),以及异构数据驱动的方法,包括DTiGEMS+(AUPR=0.957)、Trimodel(AUPR=0.946)和KGE_NFM (AUPR=0.961),都有着较好的预测性能。当数据集不平衡时,基于特征的方法和异构数据驱动的方法的 AUPR 值会不同程度地降低,但前者下降超过 10%,而后者表现更加稳定,减少约 5%。这些结果表明,基于特征的方法在应用于不平衡的数据集时容易受到影响,而异构数据驱动的方法可以部分克服这一局限性。至于端到端方法,与平衡情况相比,可能是由于不平衡情况下数据量明显增加,因此其预测性能显著提高(AUROC 约 10%,AUPR 约 9%)。这种现象表明,端到端的方法受到现有数据量的限制。因此,它们更适合大规模DTI预测。
药物冷启动的情况下,作者发现KGE_NFM(AUROC=0.853,AUPR=0.521)在AUROC方面表现最好,而RF(AUROC=0.832,AUPR=0.561)在AUPR方面表现最好。在蛋白质冷启动的情况下, KGE_NFM明显优于所有其他方法,AUPR的领先幅度显著达到19%。在基于特征的方法中,相比于RF,NFM预测性能大幅提高(在 AUROC 和 AUPR 方面约为 30%)。这一结果突出了NFM在药物-蛋白相互作用中捕捉内在联系的潜在能力,这为NFM在蛋白质的冷启动情况进行DTI预测下提供了巨大的优势。此外,通过将异构信息与传统特征相结合,KGE_NFM预测性能得到了进一步提高,AUROC为13.5%,AUPR为21%,这表明KGE提取的异构信息对于蛋白质冷启动的情况等DTI预测非常有效。

BioKG结果评价
当KG和DTI数据集中的数据量增加时,三个测试场景下的结果与之前略有不同,尤其是端到端的方法。在热启动的情况下,DeepDTI(AUROC=0.988,AUPR=0.907)表现最好,KGE_NFM(AUROC=0.987,AUPR=0.898)表现次之。在药物冷启动的情况下,基于分子指纹和蛋白质描述的传统方法RF(AUROC=0.971,AUPR=0.891)优于所有其他方法。这种现象也在其他两个基准数据集(Luo’s dataset和 Hetionet)上被观察到。此结果表明,在药物冷启动的情况下,使用基于特征的简单方法(如 RF)可能就足够了(更具体地说,是大规模虚拟筛选)。在蛋白质冷启动的情况下,KGE_NFM (AUROC=0.899, AUPR=0.549) 优于另一种异构数据驱动方法 Trimodel,AUPR 提升了15.7%。一个有趣的发现是,与Yamanishi_08的数据集相比,BioKG 数据集的端到端方法的性能有了很大的提高。例如,在药物冷启动的情况下,MPNN_CNN (AUPR=0.194) 与Yamanishi_08数据集中的RF (AUPR=0.561) 相比表现不佳。但在 BioKG 数据集中,MPNN_CNN实现了0.871的AUPR,仅比RF(AUPR=0.891) 低 2%。同样,在蛋白质冷启动的情况下,DeepDTI (AUPR=0.099)在Yamanishi_08的数据集中的表现与RF(AUPR=0.117)都很差,但在BioKG 数据集,DeepDTI(AUPR= 0.341)的表现比RF(AUPR=0.132)好很多。这一发现表明了数据集大小对端到端方法的影响,在训练集中尽可能地包含更多数量的药物和蛋白质会使端到端方法具有更好的性能。














 2972
2972











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









