






 本文详细介绍使用Quantum ESPRESSO进行自洽计算、带结构计算及FatBand分析的步骤。包括配置文件详解、计算流程指导及画图技巧,特别强调了如何选择合适的能带范围与状态组。
本文详细介绍使用Quantum ESPRESSO进行自洽计算、带结构计算及FatBand分析的步骤。包括配置文件详解、计算流程指导及画图技巧,特别强调了如何选择合适的能带范围与状态组。
1.自洽计算
复制计算后的文件到当前文件夹
cp …/phonon/pb.save/charge-density.dat pb.save/
cp …/phonon/pb.save/data-file-schema.xml pb.save/
2.band.in
/pw.x -nk 4 -in pb.band.in > pb.band.out
&control
calculation='bands'
restart_mode='from_scratch',
prefix='pb',
pseudo_dir = '../',
outdir='./'
/
&system
ibrav= 2,
celldm(1) = 9.2225583816,
nat= 1,
ntyp= 1,
ecutwfc = 30.0
occupations='smearing',
smearing='marzari-vanderbilt',
degauss=0.05
nbnd=10
/
&electrons
conv_thr = 1.0d-10
mixing_beta = 0.7
/
ATOMIC_SPECIES
Pb 207.2 pb_s.UPF
ATOMIC_POSITIONS
Pb 0.00 0.00 0.00
K_POINTS crystal_b
#这里是高对称路径
7
0.000 0.000 0.000 30
0.500 0.000 0.500 30
0.500 0.250 0.750 30
0.500 0.500 0.500 30
0.375 0.375 0.750 30
0.000 0.000 0.000 30
0.500 0.500 0.500 30
注:nbnd选择
以scf.out中这个量为基准
number of electrons = 8.00
绝缘体取1/2,金属在绝缘体的基础上加20%,加空带会拖慢计算速度,但有利于完整能带结构。
PS.这个量是怎么来的呢?
赝势文件打开搜索Z valence
所有原子的价电子加起来就是它。
3.bands.in
/bands.x -in pb.bands.in > pb.bands.out
&bands
prefix = ’pb’
outdir = ’out/’
filband = ’bands.dat’
lsym = .false.
/
4.画图
qe/plotband.x
直接在主节点运行,回答它的问题
$ ~/soft/q-e-qe-6.7MaX-Release/bin/plotband.x
Input file > bands.dat
Reading 10 bands at 251 k-points
file with representations not compatible with bands
Range: -25.4690 21.7530eV Emin, Emax, [firstk, lastk] > -26,22
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.0000
high-symmetry point: 0.0000 0.2802 0.0000 x coordinate 0.2802
high-symmetry point: 0.1401 0.1401 0.0000 x coordinate 0.4784
high-symmetry point: 0.1401 0.1401 0.1401 x coordinate 0.6185
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.8612
high-symmetry point: 0.1401 0.1401 0.0000 x coordinate 1.0594
output file (gnuplot/xmgr) > plot.band.dat
bands in gnuplot/xmgr format written to file CaH6.band.dat
最后把plot.band.dat拖去画图,注意,y列数据要减去费米能的值
费米能的值在scf.out里找
the Fermi energy is 14.5455 ev
大功告成
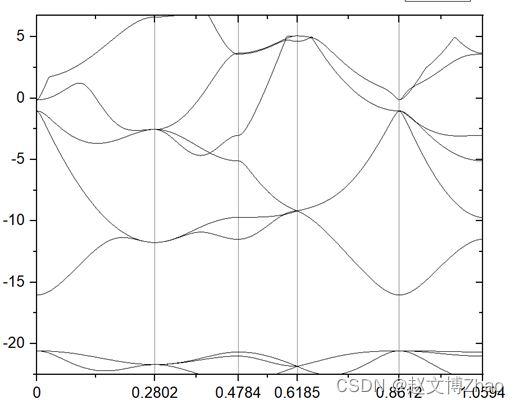
5. FatBand 分析
https://blog.csdn.net/weixin_45973682/article/details/108194472
projwfc.fat.in
&projwfc
outdir='./'
prefix='pwscf'
lsym=.false.
filproj = 'fatband'
/
运行
projwfc.x < projwfc.fat.in > projwfc.fat.out
画图 复制下面文件到计算文件夹
DFTtoolbox-master/examples/qe/soc/qe_pp.py
from DFTtoolbox.qe import postproc
import os
# Parameter ========================================================
run_task=[3,4] !选择计算内容,画胖带分析就选3,4
wkdir=os.path.dirname(os.path.realpath(__file__))
# band_read & fatband_read
Ef=17.2829 !scf.out里有,这是Fermi能
#band_plot
kdiv=[50,50,50,50,50,50] !这是高对称点插值,和band.in保持一致
klabel=['$\Gamma$','H','N','P','$\Gamma$','N']
Ebound=[-26,10] !这是你想画的图的范围,和文献对应
#fatband_plot
state_grp=[[1],[2]] 这个量的选择看后面,详细说
# Main ================================================================
pp=postproc(wkdir)
for task in run_task:
if task==1: #'band_read':
pp.band_read(Ef=Ef,bandfile='bands.dat')
elif task==2: #'band_plot':
pp.band_plot(kdiv=kdiv,klabel=klabel,Ebound=Ebound)
elif task==3: #'fatband_read':
pp.fatband_read(Ef=Ef,projout='proj.fat.out',projprefix='fatband')
elif task==4: #'fatband_plot':
pp.fatband_plot(state_grp=state_grp,kdiv=kdiv,klabel=klabel,Ebound=Ebound)
elif task==5: #pdos_read:
pp.pdos_read(Ef=Ef)
elif task==6:
pp.pdos_plot(state_grp=state_grp,Ebound=Ebound)
注意!使用的赝势要有<PP_PSWFC>部分才可以
**【state_grp】**的选择
先随便写运行一下
会得到下面输出
=> check fatband-state.dat for state_info:
[' 0 => H-1 orb-1 ( 1 / 0 / 1 / +1)\n'
' 1 => H-2 orb-1 ( 2 / 0 / 1 / +1)\n'
' 2 => H-3 orb-1 ( 3 / 0 / 1 / +1)\n'
' 3 => S-4 orb-1 ( 4 / 0 / 1 / +1)\n'
' 4 => S-4 orb-2 ( 4 / 1 / 1 / +1)\n'
' 5 => S-4 orb-2 ( 4 / 1 / 2 / +1)\n'
' 6 => S-4 orb-2 ( 4 / 1 / 3 / +1)\n']
我这是H3S的例子,H的赝势文件中PP_PSWFC里有1s轨道,S的赝势文件中PP_PSWFC里有3s,3p轨道。
所以,你想看H对能带的贡献就state_grp=[[0,1,2]]

想看S的3s轨道就state_grp=[[3]],想看S的3p轨道就state_grp=[[4,5,6]]
当然,这三张图可以写在一起state_grp=[[0,1,2],[3],[4,5,6]]


















 3985
3985

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









