







论文题目:AdsorbML: a leap in efficiency for adsorption energy calculations using generalizable machine learning potentials
Abstract:计算催化在各种应用的催化剂设计中发挥着越来越重要的作用。许多计算方法的一个常见任务是需要准确计算感兴趣的吸附物和催化剂表面的吸附能。传统上,低能吸附质-表面构型的识别依赖于启发式方法和研究人员的直觉。随着进行高通量筛选的愿望增加,单独使用heuristics(启发式)和intuition(直觉)变得具有挑战性。在本文中,我们证明了可以利用机器学习潜力来更准确、更有效地识别低能吸附物表面构型。我们的算法提供了准确性和效率之间的一系列权衡,其中一个平衡选项在87.36%的情况下都能找到最低能量配置,同时实现约 2000 倍的计算加速。为了标准化基准测试,我们引入了 Open Catalyst Dense 数据集,其中包含近 1000 个不同的表面和约 100,000 个独特的配置。
Introduction
设计新型异质催化剂在合成日常燃料和化学品中扮演着至关重要的角色。为了满足不断增长的能源需求,同时对抗气候变化,高效、低成本的催化剂对于利用可再生能源至关重要。鉴于材料设计空间的巨大,高效的筛选方法非常受欢迎。计算催化提供了筛选大量材料的潜力,以补充更耗时和成本高昂的实验研究。
许多基于第一性原理的异质催化剂发现方法的一个关键任务是计算吸附能(adsorption energy)。吸附能是与催化剂表面相互作用的分子或吸附物所关联的能量。吸附物通常被选择来捕捉反应路径中的各个步骤或中间体(例如,在CO2还原中的*CHO)。通过寻找使结构整体能量最小化的吸附物-表面构型来计算吸附能。因此,吸附能是所有潜在吸附物位置和构型的全局最小能量。这些吸附能是计算自由能图的起点,以确定催化剂表面上最有利的反应路径。已经证明,反应中间体的吸附能与实验结果(如活性或选择性)具有很强的关联性。这种从第一性原理预测催化属性趋势的能力是高效催化剂筛选方法的基础。
(ps:吸附能是指分子或原子(称为吸附物)与固体表面(称为吸附剂)之间的相互作用能量。当吸附物与吸附剂表面接触时,它们之间会产生物理或化学作用力,导致吸附物附着在表面上。吸附能就是描述这种吸附作用强度的一个物理量。)
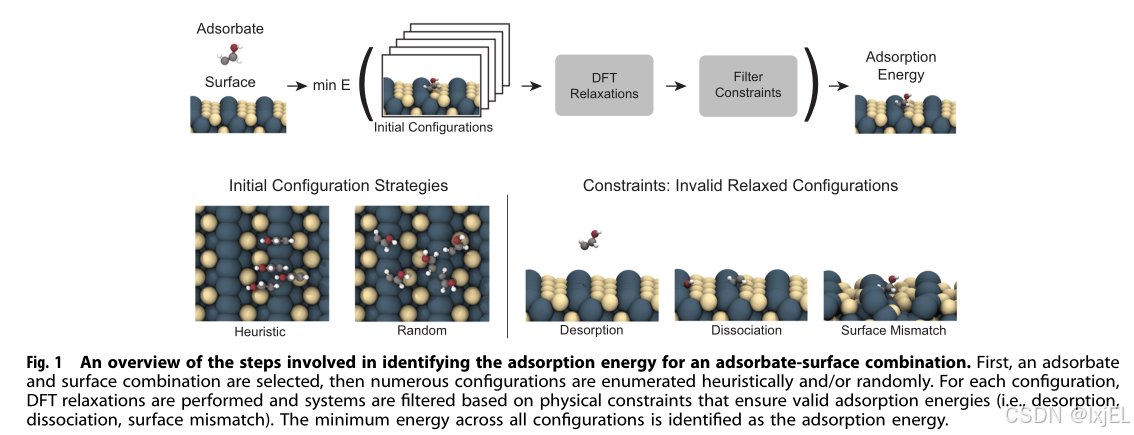
寻找吸附能呈现出许多复杂性。在表面上,吸附物有许多潜在的结合位点,对于每个结合位点,吸附物有多种取向方式(Fig.1左下角)。当吸附物放置在催化剂表面时,吸附物和表面原子会相互影响。为了确定特定吸附物-表面构型的吸附能,需要放松原子位置,直到达到局部能量最小值。密度泛函理论(DFT)是执行这种吸附物-表面松弛的最常用方法。DFT首先进行单点计算,输出是系统的能量和每个原子的力。然后进行松弛,进行局部优化,其中每个原子的力通过DFT迭代计算,并使用优化算法(例如,共轭梯度)更新原子位置,直到找到局部能量最小值。为了找到全局最小值,需要一种采样吸附物-表面构型的策略和/或一种技术,例如在优化过程中克服能量障碍的最小跳跃,这需要策略。
吸附能(
△
E
a
b
s
\bigtriangleup E_{abs}
△Eabs)的计算是吸附表面的能量
E
s
y
s
E_{sys}
Esys减去清洁表面的能量
E
s
l
a
b
E_{slab}
Eslab和气相吸附物或参考物质的能量
E
g
a
s
E_{gas}
Egas。
△
E
a
b
s
=
E
s
y
s
−
E
s
l
a
b
−
E
g
a
s
(
1
)
\bigtriangleup E_{abs}=E_{sys}-E_{slab}-E_{gas}(1)
△Eabs=Esys−Eslab−Egas(1)
松弛的吸附物-表面结构必须遵守某些期望的属性,以便它们的吸附能量既准确又有效。一个约束的例子是,在最终松弛结构中,吸附物不应被解吸,即不应从表面浮起(Fig.1右下角)。此外,如果吸附物有多个原子,它不应解离或分裂成多个吸附物,因为它将不再是感兴趣的分子的吸附能量。类似地,如果吸附物与清洁表面相比在表面上引起显著变化, E s l a b E_{slab} Eslab将造成表面不匹配。重要的是要注意,如果一个松弛结构违反了这些约束之一,并不一定意味着松弛是不准确的;这些结果确实会出现,但它们会导致无效或不准确的吸附能量,因为它已经被定义。
(ps:松弛的吸附物通常指的是在吸附过程中,由于吸附剂表面的力场作用,吸附物分子会发生位置、构型或者状态的变化,最终达到一个相对稳定的状态。这个过程称为吸附物的松弛。松弛过程可能包括吸附物分子在表面的扩散、吸附物分子构象的变化、吸附层厚度的增加等。在物理吸附中,松弛通常与吸附剂表面的范德华力有关,而在化学吸附中,可能涉及到化学键的形成和断裂)
识别全局最优的吸附物-表面构型历来依赖于专家直觉或更近期的启发式方法。直觉和试错法可以用于一次性的系统,但它不适用于大量系统。常用的启发式方法通常基于表面对称性。这些方法在过去基于描述符的研究中已经成功应用。最近,一种基于图的方法被用来识别独特的吸附物-表面构型。然而,随着表面和吸附物的复杂性增加,寻找最低能量吸附物-表面构型挑战性大大增加。当吸附物是柔性的,拥有多种自身的配置时,这种情况尤其具有挑战性,因为系统中有许多有效的自由度。
虽然密度泛函理论(DFT)能够准确估计原子力和能量,但它在计算上非常昂贵,随着电子数量的增加而呈O(N^3)的规模增长。使用完整的DFT松弛来评估单一的吸附物-表面构型可能需要大约24小时来计算。由于通常需要探索大量的构型来找到吸附能,所有涉及的DFT计算可能需要几天甚至几周的时间。假设有人想要通过使用5个吸附物描述符,每个材料约90个表面,每个表面约100个位点,从材料项目数据库中暴力筛选10万种材料用于二氧化碳还原反应(CO2RR),那么将需要大约45亿CPU天的计算量,这对于世界上最大的超级计算机来说也是一个难以解决的问题。为了显著减少所需的计算量,一个有前景的方法是使用机器学习势能来加速寻找最低能量吸附物-表面构型的搜索。
最近,用于估算原子力和能量的机器学习(ML)势能在标准基准测试中显示出了显著的进展,同时比密度泛函理论(DFT)快好几个数量级。尽管在大型且多样的Open Catalyst 2020 数据集(OC20)上,ML在放松能量估算上的准确性已经提高到了0.3电子伏特,但为了准确筛选,仍然希望达到0.1电子伏特的准确性。这就提出了一个问题,即是否可以通过结合使用DFT和ML势能的混合方法来实现高准确性,同时保持效率。
在没有标准化验证数据的情况下,评估寻找低能吸附物-表面构型新方法的性能是具有挑战性的。通常,新方法会在相对较少的系统上进行测试,这使得评估泛化能力变得困难。虽然Open Catalyst 2020 数据集(OC20)包含了大约100万个“吸附能”,但它并没有对每个吸附物-表面组合进行多重配置的采样,这意味着被放松的那一个构型不太可能是全局最小值。这使得OC20成为一个不适用于寻找最小结合能的数据集。为了解决这个问题,我们引入了Open Catalyst 2020-Dense 数据集(OC20-Dense)。OC20-Dense包括两个部分——一个验证集和一个测试集。验证集用于开发;测试集用于报告性能。每个部分包含了大约1000个来自OC20数据集验证和测试集的独特吸附物-表面组合。OC20-Dense中的任何数据都不会用于训练。为了探索我们方法的泛化能力,我们从OC20的四个子集中各取了大约250个组合——领域内(ID)、领域外(OOD)-吸附物、OOD-催化剂和OOD-两者。对于每个组合,我们执行了密集的初始构型采样,并使用DFT计算放松,以创建一个强有力的基线,用于评估估算的吸附能。
我们提出了一种估算吸附能的混合方法,充分利用了机器学习(ML)势能和密度泛函理论(DFT)的优势。我们使用启发式和随机策略来采样大量潜在的吸附物构型,并使用ML势能进行放松。然后,可以使用单点DFT计算或完整的DFT放松来细化最佳的k个放松能量。通过这种方法,可以在准确性和效率之间做出适当的权衡。
大量的研究工作一直致力于通过改进初始结构生成和全局优化策略来确定最低能量的吸附物-表面构型。Peterson采用了最小跳跃法,并开发了一种使用约束最小跳跃的全局优化方法来保持吸附物的身份。然而,该方法完全依赖于DFT来执行搜索,这使得它在计算上仍然很昂贵。最近,Jung等人提出了一个主动学习工作流程,其中使用高斯过程来运行约束最小跳跃模拟。他们的模拟生成的结构通过DFT进行验证,并迭代地添加到训练集中,直到模型收敛。然后训练好的模型并行运行约束最小跳跃模拟,一部分结构通过DFT进行细化,最终确定吸附能。我们注意到,以往使用机器学习模型来加速这一过程的尝试通常依赖于针对每种吸附物/催化剂组合的定制模型,这限制了更广泛的适用性。在继续降低人力和计算成本的同时,极大地扩展这些方法的多功能性的一种可能性是使用可泛化的机器学习势能来加速寻找低能量吸附物-表面构型。
这项工作的贡献有三个方面:
- 我们提出了AdsorbML算法,以在准确性和效率之间进行权衡,识别吸附能。
- 我们开发了Open Catalyst 2020-Dense数据集(OC20-Dense),以基准测试吸附能搜索任务。
- 我们使用提出的AdsorbML算法在OC20-Dense上对文献中的图神经网络(GNN)模型进行了基准测试;识别出几个适合实际筛选应用的有前景的模型。
Result
OC20-Dense evaluation
为了评估计算吸附能的方法,我们提出了Open Catalyst 2020-Dense数据集(OC20-Dense),该数据集通过密集探索每个独特的吸附物-表面系统的众多构型,紧密近似于真实吸附能。每个OC20-Dense的分割包含约1000个独特的吸附物-表面组合,涵盖74种吸附物、800多个无机块体晶体结构,以及总共超过80,000个通过启发式和随机生成的构型。Table 1提供了两个分割的总结。完成该数据集需要大约400万CPU小时的计算。关于OC20-Dense的更详细讨论可以在 Method 部分找到。
我们报告了在OC20上先前基准测试的一系列图神经网络(GNN)的结果,以评估现有模型在OC20-Dense上的性能。这些模型包括SchNet、DimeNet++、PaiNN、GemNet-OC、GemNet-OC-MD、GemNet-OC-MD-Large、SCN-MD-Large和eSCN-MD-Large,其中MD表示在OC20及其伴随的从头算分子动力学(MD)数据集上进行训练。这些模型并非作为本工作的一部分进行训练;训练好的模型直接取自先前发表的工作,可以在Open Catalyst Project找到。在这些模型中,eSCN-MD-Large和GemNet-OC-MD-Large目前在OC20和Open Catalyst 2022数据集(OC22)上都是表现最好的模型。探索这些趋势在OC20-Dense上的表现将很重要,以了解在OC20上取得的进展如何转化为这里提出的更重要的下游任务。
理想情况下,OC20-Dense的基准真值将是每个吸附物-表面系统所有可能构型中的最低放松能量。由于可能的构型数量是组合性的,社区已经开发了启发式方法来放置催化剂表面上的吸附物。当我们仅评估启发式构型时,我们将其称为DFT-Heuristic-Only (DFT-Heur)。为了增加构型空间,我们还在表面上均匀随机采样位点,并将吸附物随机旋转放置在这些位点上,同时在z轴上进行随机旋转,在x和y轴上轻微摆动。当我们针对启发式和随机构型进行评估时,我们将其称为DFT-Heuristic-Only (DFT-Heur)。尽管计算上更加昂贵,但这个基准提供了对构型的更全面搜索,并且比仅使用启发式构型提供了更准确的吸附能估计,后者是社区常用的基准。关于这两个基准的更多细节可以在“方法”部分找到。
ML relaxations
我们探索了机器学习(ML)预测能在多大程度上找到在DFT最小能量阈值内或更低的吸附能。虽然一个完美的ML替代DFT只能匹配DFT,但力的微小误差和优化器的差异有可能给放松过程增加噪声,并导致之前未探索的构型。对于每个模型,都在相同的一组吸附物构型上执行放松。初始构型是基于文献中常用的启发式策略和在表面上随机生成的构型创建的。对所有初始构型进行ML驱动的放松;由于物理约束不适合进行吸附能计算的系统被移除,包括解离、脱附和表面不匹配。关于放松约束的深入讨论可以在Method部分找到。
在评估性能时,我们定义成功为在OC20-Dense中找到的吸附能与DFT吸附能相差在可接受的容差内(本研究中为0.1电子伏特)或更低。需要注意的是,OC20-Dense中的基准真值吸附能是一个上限,因为可能存在更低的吸附能。在评估ML预测的吸附能时,必须使用单点DFT计算来验证结果,因为没有下限的评估指标很容易被预测低能量所操纵(详见补充信息SI)。为了可靠地评估ML,我们认为如果ML吸附能与DFT吸附能相差0.1电子伏特或更低,并且对应的预测ML结构的DFT单点评估与预测的ML能量相差0.1电子伏特或更低,则ML吸附能是成功的。这确保了ML预测不仅找到了低吸附能,而且准确且不是人为夸大的。结果报告在Table 2中,其中顶级的OC20模型,包括eSCN-MD-Large和GemNet-OC-MD-Large,分别达到了56.52%和48.03%的成功率。表2还报告了ML和DFT吸附能之间的能量平均绝对误差(MAE),与成功率和OC20的S2EF指标很好地相关。
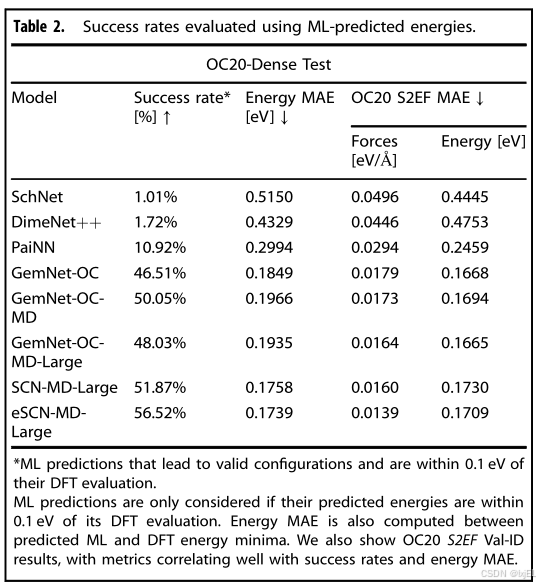
虽然目前的模型已经取得了令人难以置信的进步,但对于日常实践者来说,仍需要更高的成功率。在高通量筛选的环境中,成功的候选者将进入更昂贵的分析甚至实验合成,大约50%的成功率可能导致在研究假阳性上浪费大量的时间和资源。随着模型开发将继续帮助改进指标,这项工作探索了混合ML+DFT策略,以额外的计算成本为代价来提高成功率。
AdsorbML algorithm
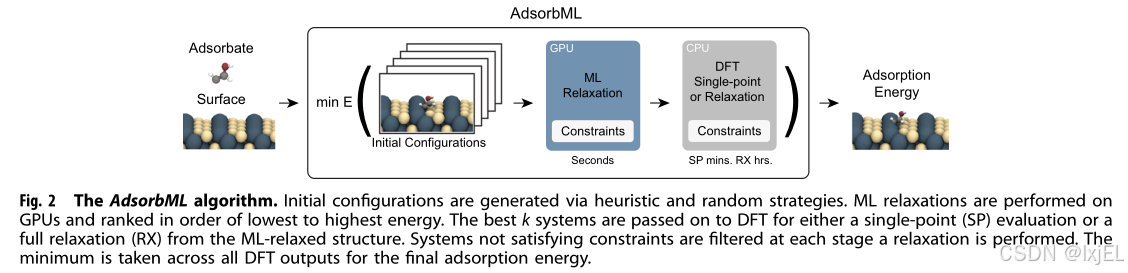
我们引入了AdsorbML算法,使用机器学习(ML)来加速吸附物放置过程(见Fig 2)。对于每个模型,我们探索了两种结合ML的策略,随后进行DFT计算以确定吸附能。我们注意到这种策略是通用的,可以与任何初始构型算法一起使用。
在这两种方法中,第一步是生成ML放松。然而,我们并不是简单地取ML放松能量的最小值,而是根据能量从低到高对系统进行排序。选择能量最低的k个系统,然后(1)对相应的结构进行DFT单点计算(ML+SP)或(2)从ML放松的结构进行DFT放松(ML+RX)。第一种策略旨在获得ML预测放松结构的更可靠能量测量,而第二种策略将ML视为预优化器,DFT完成放松。通过选择能量最低的k个系统,我们为模型提供了k次机会来达到可接受的准确吸附能。随着k的增加,涉及更多的DFT计算,但与完整的DFT方法相比,我们仍然预期会有显著的节省。特定系统的吸附能是通过取最佳k个DFT后续计算的最小值来获得的。
在这两种策略中,ML能量仅用于对构型进行排序,最终的能量预测来自DFT计算。虽然从计算角度来看,完全依赖ML是理想的,但使用DFT既可以提高准确性,又提供了一个验证步骤,使我们对吸附能预测更有信心。
Experiments
我们的目标是在OC20-Dense中找到与单独使用DFT相当或更好的吸附能。我们用来量化这项任务的指标是成功率,即我们的ML+DFT吸附能与DFT吸附能相差0.1电子伏特或更低的OC20-Dense系统中的百分比。这些实验中不包括ML能量的验证,因为所有最终的吸附能至少来自一次DFT计算,确保所有值都是有效的。另一个我们跟踪的指标是与DFT-Heur+Rand基线相比的加速比。加速比是按DFT-Heur+Rand使用的DFT电子步骤与提出的混合ML+DFT策略的比率来评估的。关于这些指标的更详细讨论可以在Method部分找到。除非另有说明,所有结果都在测试集上报告,验证集上的结果可以在补充信息(SI)中找到。在评估仅使用DFT计算的常见基线DFT-Heur时,以1.81倍的加速比实现了87.76%的成功率。
ML+SP(single-point)
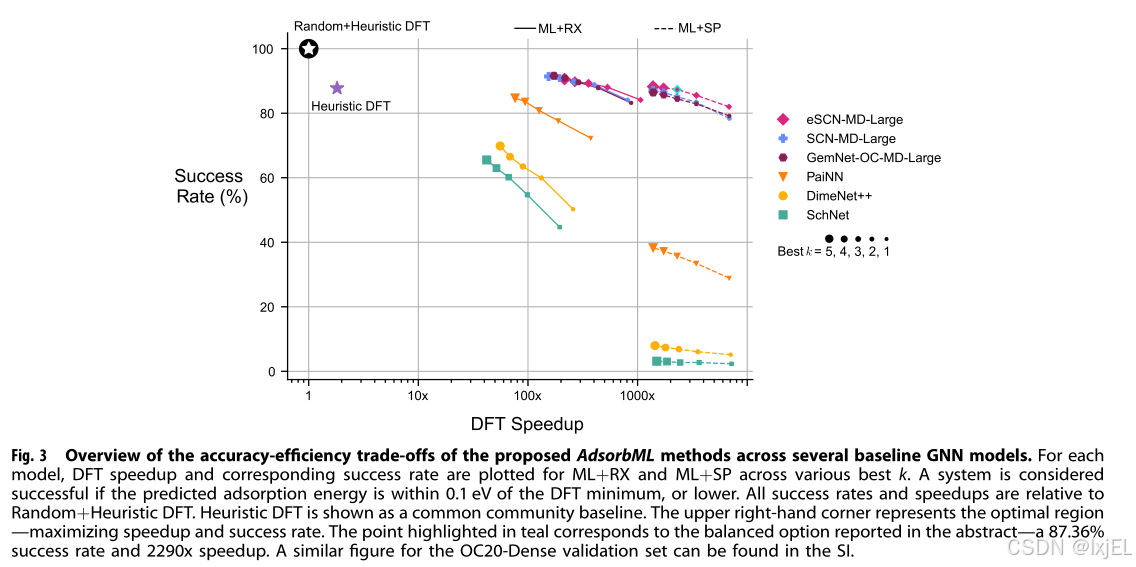
在ML放松状态上使用单点评估的结果总结在Fig 3中。eSCN-MD-Large和GemNet-OC-MD-Large在k=5时实现了86%以上的成功率,其中eSCN-MD-Large以88.27%的成功率超越了所有模型,略优于DFT-Heur基线。其他模型,包括SchNet和DimeNet++,成功率低至3.13%和7.99%,表明预测的放松结构非常不利。所有模型的加速比相当可比,对于k=5,加速比在1400倍到1500倍之间,比DFT-Heur基线快几个数量级。具体来说,eSCN-MD-Large和GemNet-OC-MD-Large分别提供了1384倍和1388倍的加速比。如果速度是最重要的,即使在保持eSCN-MD-Large 82%的成功率的同时,也能达到6817倍的加速比。在更平衡的权衡点,k=3时,eSCN-MD-Large和GemNet-OC-MD-Large的成功率分别为87.36%和84.43%,同时保持了2296倍和2299倍的加速比。在Fig 4中,比较了不同模型使用ML+SP识别的几个系统的最小能量结合位点。

ML+RX(relaxed structures)
单点评估提供了对ML结构的快速评估,但性能严重依赖于预测放松结构的准确性。这一点在用DFT评估ML放松结构的最大每个原子力范数时尤为明显。SchNet和DimeNet++平均最大力分别为2.00 eV/Å和1.21 eV/Å,进一步支持了这些模型在获得有效放松结构方面面临的挑战。另一方面,像GemNet-OC-MD-Large和eSCN-MD-Large这样的模型平均最大力分别为0.21 eV/Å和0.15 eV/Å。虽然这些模型更接近于有效的放松结构(即最大力≤0.05 eV/Å),但这些结果表明仍有进一步优化的空间。从ML+RX进行DFT放松的结果在Fig 3中绘制。eSCN-MD-Large和GemNet-OC-MD-Large在所有k值上都超越了所有模型,分别为90.60%和91.61%的成功率。鉴于与ML+RX相关的额外DFT成本,加速比不出所料地降低了。在k=5时,eSCN-MD-Large和GemNet-OC-MD-Large的加速比分别为215倍和172倍。SchNet和DimeNet++的加速比要小得多,分别为42倍和55倍。与SchNet和DimeNet++相关的较小加速比表明,需要更多的DFT步骤来放松模型生成的先前不利配置。相反,eSCN-MD-Large的较大加速比可以归因于其预测中几乎放松的状态(平均最大力约为0.15 eV/Å)。在k=1时,eSCN-MD-Large仍能保持84.13%的成功率,加速比高达1064倍。在更平衡的权衡点,即k=3时,eSCN-MD-Large和GemNet-OC-MD-Large的成功率分别为89.28%和89.59%,同时保持了356倍和288倍的加速比。
结果表明,在准确性和效率之间存在一系列的权衡,这是在选择策略时应考虑的。对于我们最好的模型,ML+SP的结果几乎比ML+RX快8倍,成功率仅略有下降(3-4%),表明这是一个值得的妥协。对于表现较差的模型,这种差异更为显著。
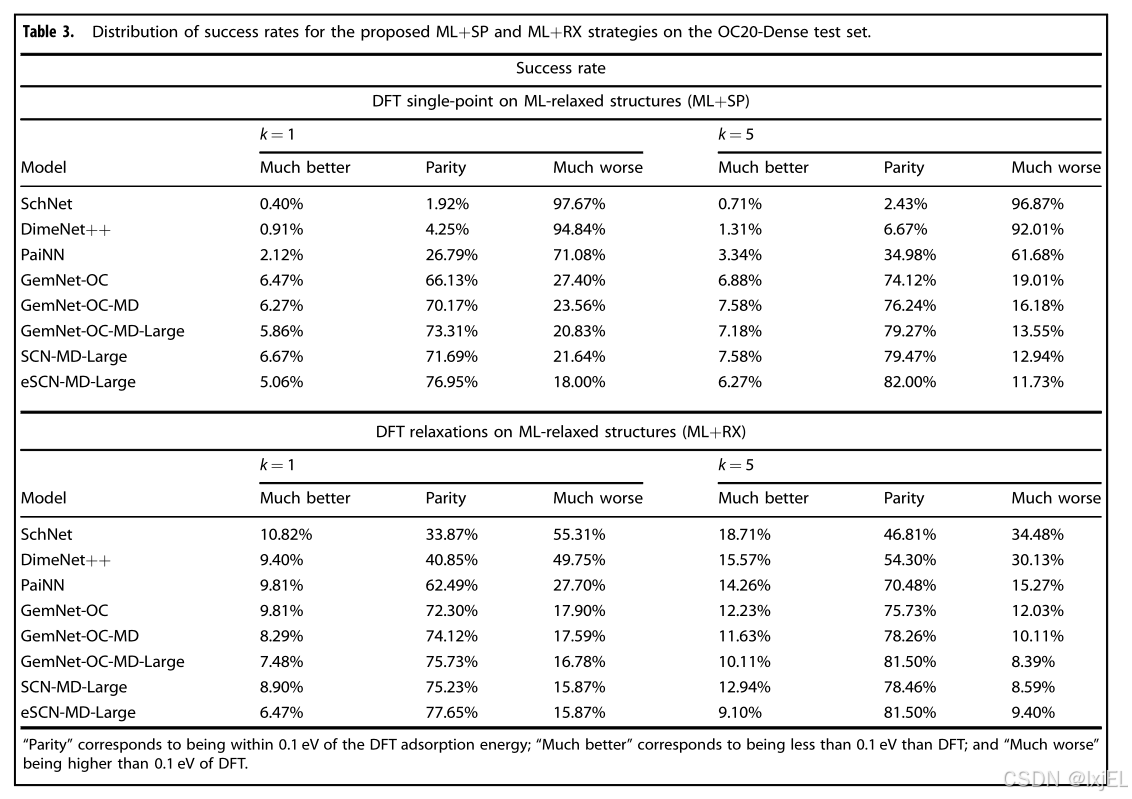
在表3中,我们测量了预测分布与基准真值相比好得多、相当或差得多的情况,其中“好得多/差得多”对应于比DFT吸附能低/高0.1电子伏特。在两种策略中,我们观察到最准确的模型并不一定能找到好得多的最小值。例如,在k=5的ML+RX中,eSCN-MD-Large发现有9.10%的系统具有好得多的最小值,而DimeNet++发现有15.57%。同样,尽管eSCN-MD-Large在ML+SP中超越了其他模型,但在ML+RX中观察到的改进较小;这是模型到达一个相当大的局部最小值的后果,随后的DFT放松几乎没有好处。这进一步表明,模型中的某种噪声可能有助于找到更好的最小值。ML+SP和ML+RX实验的完整结果集可以在补充信息(SI)中找到,包括OC20-Dense测试集和验证集的结果。
Distribution splits
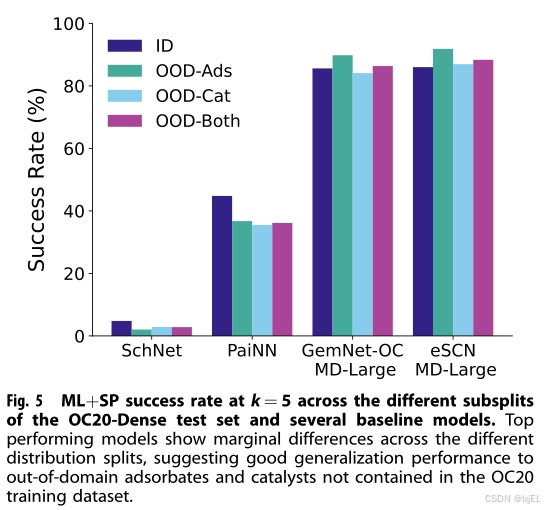
此外,我们评估了在不同数据子集上的成功率指标。OC20-Dense从四个OC20子集—ID(领域内)、OOD-Adsorbate(领域外-吸附物)、OOD-Catalyst(领域外-催化剂)和OOD-Both(领域外-两者)中均匀采样。在我们最好的模型中,我们观察到在不同分布子集上的性能保持一致(Fig 5)。这表明,对于包括不在OC20中的吸附物或表面的应用,AdsorbML仍然提供准确和有意义的结果。虽然我们预期结果将与OC20一致,即ID的表现优于OOD,但这里并不一定如此。例如,eSCN-MD-Large在k=5的ML+SP中,在ID上达到86.00%的成功率,而在OOD-Both上达到88.35%的成功率,ML+RX也有类似的趋势。我们认为这种差异是由于每个子集中的样本量相对较小(250个)。完整的结果集可以在补充信息(SI)中找到。
Configuration analysis
在主要结果之外,我们还探索了仅使用启发式或仅使用随机ML构型在OC20-Dense验证集上的性能。报告的结果是基于SCN-MD-Large模型,采用ML+SP策略。当k=5时,仅使用随机构型,成功率从87.77%下降到82.94%。更显著的是,当仅考虑启发式构型时,成功率大幅下降至62.18%。这表明随机构型可能具有更大的影响。额外的结果可以在补充信息(SI)中找到。
Discussion
我们将这项工作视为减少DFT计算成本的重要但初步的一步,这不仅适用于催化应用,而且更广泛地适用于计算化学领域。AdsorbML提供了一个准确性与效率之间的权衡选择谱,可以根据应用和可用的计算资源来选择。例如,如果我们希望在固定的计算预算下筛选尽可能多的二氧化碳还原反应催化剂,我们可以选择k=2的ML+SP策略,成功率为85%,同时筛选的材料数量是单独使用DFT时的约3400倍。另一方面,如果研究的深度更为重要,ML+RX是一个不错的选择,因为结构是完全用DFT优化的,计算加速来自于减少所需的总放松步骤。在这种情况下,ML势能作为一个高效的预优化步骤。尽管ML模型占整体计算的一小部分(详见SI中的详细信息),但我们预计随着未来对推理效率的更多关注,这些需求将进一步降低。
一个值得进一步研究的观察是,机器学习(ML)模型在5%到15%的时间里找到了更好的最小值,这取决于效率权衡(Table 3)。如果我们的ML模型是完美的,那么就不会有更低吸附能的情况;然而,以不准确力预测形式的隐含噪声允许ML模型穿越势能表面的未探索区域。探索隐含和显式噪声在多大程度上影响ML放松和下游任务(如成功率)是一个重要的未来研究方向。
这项工作的另一个自然延伸是关注全局优化和初始构型生成的替代方法。在这里,我们专注于通过枚举初始吸附物-表面构型来加速寻找全局最小值的蛮力方法。然而,可能存在更有效的全局优化方法,如最小跳跃、约束优化、贝叶斯优化或直接学习的方法。值得注意的是,尽管我们的枚举覆盖了比传统启发式方法更大的空间,但它并不是穷尽和全面的。我们发现,增加超过已采样数量的随机构型数量,其回报逐渐减少,因为从使用启发式+80%随机构型DFT到使用启发式+100%随机构型DFT的成功率变化仅为1.6%(更多细节请参见SI)。如果筛选更多的机器学习配置仍然是有利的,那么考虑我们如何处理重复的结构可以进一步提高准确性和效率。我们在SI中简要探讨了这一点,其中去除具有几乎相同ML能量的系统会产生边际效益。
虽然像GemNet-OC和eSCN-MD-Large这样的当前模型在OC20-Dense上展示了令人印象深刻的成功率,但不经过后续DFT的ML放松仍然不够准确,无法满足实际应用的需求(Table 2)。为了让未来的模型开发工作能够应对这一挑战,值得强调一些观察结果。首先,OC20-Dense上的成功率与S2EF和基于放松的初始结构到放松能量(IS2RE)OC20任务之间存在正相关。因此,基于放松的IS2RE和S2EF指标可以作为在OC20上训练模型时的代理。关于模型开发的另一个重要注意事项是,OC20-Dense的验证集是OC20验证集的一个子集;因此,在评估OC20-Dense时,不应使用OC20验证数据进行训练。最后,强烈建议在报告OC20-Dense验证集上的结果时,使用DFT单点计算进行评估,因为成功率指标可能通过仅预测低能量来操纵。这可以通过大约1000个单点计算来完成。随着OC20-Dense测试集的发布,我们将探索发布一个公共评估服务器,以确保DFT评估的一致性和可访问性,如果有兴趣的话。
在化学领域的数据集和机器学习方面取得了巨大进步,使得模型能够显著增强和扩充DFT(密度泛函理论)计算。我们的结果表明,当前最先进的ML(机器学习)模型不仅加速了催化过程的DFT计算,而且能够更准确地估计需要全局优化的属性,例如吸附能。虽然本工作中使用的模型最适合理想的吸附物-表面催化剂,但微调策略可以帮助扩展应用到其他化学领域,包括金属有机框架和沸石。同样,本工作中使用的模型是在一致的DFT理论水平(修订的Perdew-Burke-Ernzerhof,无自旋极化)上训练的,通过微调或其他训练策略,也可以推广到其他泛函和理论水平。考虑到ML模型开发的时间线,这些结果在几年前还不可能实现。我们预计这项工作将加速对复杂吸附物-表面构型的大规模探索,涵盖广泛的化学和应用领域。将这些结果推广到更多样的材料和分子,而不依赖于DFT,是未来社区面临的重大挑战。
Method
Open Catalyst 2020-Dense Dataset (OC20-Dense)
评估吸附能估算需要一个基准真值数据集,该数据集彻底探索了潜在的吸附构型集合。虽然OC20计算了大约100万个系统的吸附能,但这些能量可能并不对应于特定吸附物-表面组合的最小值。更具体地说,对于给定的催化剂表面,OC20考虑了所有可能的吸附位点,但只在一种特定配置中随机选择的一个位点上放置所需的吸附物。OC20提出的任务促进了更准确的机器学习势能用于催化反应的发展,但像IS2RE这样的任务,尽管相关性很好,但在评估性能时并不总是足够的,因为当模型找到不同的、更低能量的最小值时会受到惩罚——这是一个更理想的结果。作为OC20任务的自然延伸,我们引入了OC20-Dense来调查模型在寻找吸附能方面的性能。
OC20-Dense的构建旨在密切近似特定吸附物-表面组合的吸附能。为了实现这一点,需要密集采样初始吸附构型。OC20-Dense由两个分割组成——一个验证集和一个测试集。对于每个分割,从相应的OC20验证/测试集中采样了大约1000个独特的吸附物-表面组合。然后从每个子分割(ID、OOD-Adsorbate、OOD-Catalyst、OOD-Both)中均匀采样,以探索模型在这项任务上的泛化能力。对于每个吸附物-表面组合,使用了两种策略来生成初始吸附物构型:启发式和随机。启发式策略旨在代表平均催化研究者,其中使用像CatKit和Pymatgen这样的流行工具来制作初始构型。给定一个吸附物和表面,Pymatgen枚举所有对称相同的位点,将吸附物放置在位点上,并对吸附物沿z轴进行随机旋转,然后在x和y轴上应用轻微的摆动。虽然启发式策略寻求捕捉最佳实践,但它们确实限制了可能的搜索空间,并且不能保证选择的是真正的最小能量。为了解决这个问题,我们还随机枚举了表面上的M个位点,然后将吸附物放置在选定的位点上。在这项工作中,使用了M=100,并对吸附物以类似的方式进行随机旋转。在两种策略中,我们移除了不合理的构型——吸附物没有放置在板上和/或放置得过于深入表面。然后对所有构型进行DFT放松,结果过滤掉那些脱附、解离或造成表面不匹配的构型。在剩余的构型中,最低的能量被认为是吸附能。虽然随机策略意味着更全面的枚举,但它并不完美,可能会错过一些吸附物构型。OC20-Dense验证集的创建方式类似,但存在一些显著差异,详细信息在补充信息(SI)中概述。
OC20-Dense测试集包含了989个独特的吸附物+表面组合,涵盖了74种吸附物和837种块体。在进行密集采样之后,共计算了56,282个启发式和49,432个随机构型,使用了DFT。平均每个系统有56个启发式和50个随机构型(注:虽然生成了M=100个随机位点,但在筛选后可用的位点较少)。总共约使用了400万小时的计算资源来创建数据集。所有DFT计算都是使用维也纳从头算模拟包(VASP)进行的。关于DFT设置和详细信息的讨论可以在补充信息(SI)中找到。
Evaluation metrics
为了充分追踪进展,我们提出了两个主要指标——成功率和DFT加速比。成功率是策略返回的能量在DFT吸附能的σ内或更低的比例。选择了σ = 0.1电子伏特作为误差范围,因为社区通常愿意为了实际相关性而容忍一定程度的误差。一旦模型和策略达到饱和,收紧这个阈值以提高准确性是一个可预见的步骤。虽然通过增加DFT计算可以得到高成功率,但我们使用DFT加速比作为评估效率的手段。加速比是DFT-Heur+Rand使用的DFT电子或自洽(SC)步骤与所提出策略使用的步骤之比。我们使用电子步骤,因为我们发现它们与DFT计算时间的相关性比离子或放松步骤的数量更好。失败的或导致无效结构的DFT计算被包括在加速比评估中,因为它们仍然代表筛选中实现的成本。我们选择不包括计算时间在这个指标中,因为结果通常依赖于硬件,这可能会使比较结果变得不可靠。ML放松成本不包括在这个指标中,因为硬件差异以及CPU+GPU计时使其难以标准化。虽然与DFT计算相比,ML计时通常可以忽略不计,但关于ML计时的更详细分析可以在补充信息(SI)中找到。这些指标是针对严格的基准真值——DFT-Heur+Rand报告的,并与社区的启发式实践——DFT-Heur进行了比较。正式地,这些指标在方程(2)和(3)中定义。
s
u
c
c
e
s
s
r
a
t
e
=
∑
i
N
l
[
m
i
n
(
E
^
i
)
−
m
i
n
(
E
i
)
≤
σ
]
N
(
2
)
success\space rate=\frac{ {\textstyle \sum_{i}^{N}}\mathbb{l} [min(\hat{E} _{i})-min(E_{i})\le \sigma ] }{N}(2)
success rate=N∑iNl[min(E^i)−min(Ei)≤σ](2)
D
F
T
S
p
e
e
d
u
p
=
∑
N
N
S
C
s
t
e
p
s
∑
N
N
^
S
C
s
t
e
p
s
(
3
)
DFT\space Speedup=\frac{\sum_{N}N_{SCsteps}}{\sum_{N}\hat N_{SCsteps}} (3)
DFT Speedup=∑NN^SCsteps∑NNSCsteps(3)
其中
i
i
i是一个吸附物-表面系统,
N
N
N是独特系统的总数,
l
(
x
)
\mathbb{l}(x)
l(x)是指示函数,
^
\hat{}
^ 是所提出的策略,
N
S
C
s
t
e
p
s
N_{SCsteps}
NSCsteps是自洽或电子步骤的数量,
m
i
n
(
E
)
min(E)
min(E)是特定系统所有构型中的最小能量。对于这两个指标,数值越高越好。
Relaxation constraints
在我们的分析中,可能需要排除一些考虑中的吸附物-表面构型,因为它们在放松后可能进入了必须舍弃的状态。对于这项工作,我们考虑了三种这样的情况:
(1)脱附,(2)解离,以及(3)显著的吸附物诱导的表面变化。
脱附,即吸附分子不与表面结合,通常问题不大,因为脱附系统的能级通常较高。尽管如此,了解所考虑的构型实际上都没有吸附到表面上是有用的。解离,即吸附分子分解成不同的原子或分子,是个问题,因为得到的吸附能不再与我们感兴趣的一致,即单个分子的吸附能,而不是两个或更多更小分子的吸附能。包含这些系统可能会显示出更低的吸附能,但由于能量不代表所需的系统,可能会导致误报。
最后,我们还会丢弃那些有显著吸附物诱导表面变化的系统,因为与解离一样,我们不再计算感兴趣的能量。在计算吸附能时,会包括一个干净、放松表面的能级项。这种计算的一个基本假设是,相应的吸附物-表面系统的生成表面必须与相应的干净表面可比,否则这种参考方案就会失败,得到的吸附能就会不准确。
对于这些情况,我们根据邻域连通性、距离信息和原子共价半径开发了检测方法。根据用户的应用程序,可以决定是否收紧在内定义的阈值。每种检测方法的详细信息和进一步讨论可以在补充信息(SI)中找到。

















 2025
2025

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









