







题目目录
- 1. 安装一些R包。
- 2. 了解ExpressionSet对象,比如CLL包里面就有data(sCLLex),找到它包含的元素,提取其表达矩阵(使用exprs函数),查看其大小。
- 3. 了解 str,head,help函数,作用于第二步提取到的表达矩阵。
- 4. 安装并了解hgu95av2.db包,看看ls("package:hgu95av2.db")后显示的那些变量。
- 5. 理解head(toTable(hgu95av2SYMBOL))的用法,找到 TP53 基因对应的探针ID。
- 6. 理解探针与基因的对应关系,总共多少个基因,基因最多对应多少个探针,是哪些基因,是不是因为这些基因很长,所以在其上面设计多个探针呢?
- 7. 第二步提取到的表达矩阵是12625个探针在22个样本的表达量矩阵,找到那些不在hgu95av2.db包收录的对应着SYMBOL的探针。
- 8. 过滤表达矩阵,删除那942个没有对应基因名字的探针。
- 9. 整合表达矩阵,多个探针对应一个基因的情况下,只保留在所有样本里面平均表达量最大的那个探针。
- 10. 把过滤后的表达矩阵更改行名为基因的symbol,因为这个时候探针和基因是一对一关系了。
- 11. 对第10步得到的表达矩阵进行探索,先画第一个样本的所有基因的表达量的boxplot,hist,density,然后画所有样本的这些图。
写在前面——这次的R语言习题比上次的中级题目做起来舒服一些。花了四五天的时间整理了一下,因为题目太长,所以划分成两篇文章来写。不过还有地方不明白,先挖个坑,之后来填。需要代码文件的私信我即可。
题目原文:http://www.bio-info-trainee.com/3409.html
参考答案:https://www.jianshu.com/p/dd4e285665e1 https://www.jianshu.com/p/dd4e285665e1
参考答案:https://www.jianshu.com/p/c62cbb9e1a2e
下篇指路:https://blog.csdn.net/narutodzx/article/details/119775994
1. 安装一些R包。
数据包: ALL, CLL, pasilla, airway
软件包:limma,DESeq2,clusterProfiler
工具包:reshape2
绘图包:ggplot2
不同领域的R包使用频率不一样,在生物信息学领域,尤其需要掌握bioconductor系列包。
# R包的安装一般分为两种
# 1.对于bioconductor的安装。可在下面的网站中查询包是否属于bioconductor系列
# https://www.bioconductor.org/packages/release/BiocViews.html#___Software
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c('ALL','CLL','pasilla','clusterProfiler'))
BiocManager::install(c('airway','DESeq2','edgeR','limma'))
# 2.对于普通包的安装
install.packages("reshape2", "ggplot2")
2. 了解ExpressionSet对象,比如CLL包里面就有data(sCLLex),找到它包含的元素,提取其表达矩阵(使用exprs函数),查看其大小。

ExpressionSet 类旨在将多个不同的信息源组合到一个方便的结构中。
ExpressionSet 可以方便地操作(例如,子集化、复制),并且是许多 Bioconductor 函数的输入或输出。
ExpressionSet类包含:
assayData:微阵列表达数据
phenodata:实验样本的描述
featuredata:实验所用芯片或技术的特点
annotation:注释信息
experimentData:描述实验的一种灵活结构
suppressPackageStartupMessages(library(CLL))
data("sCLLex")
sCLLex
# 获得表达矩阵
exprSet <- exprs(sCLLex)
class(exprSet)
dim(exprSet)
#查看样本编号
sampleNames(sCLLex)
#查看所有表型变量
varMetadata(sCLLex)
#查看基因芯片编码
featureNames(sCLLex)[1:100]
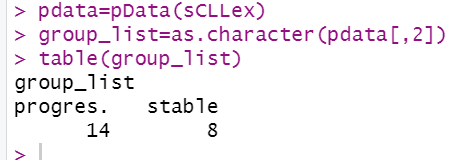
# 取sCLLex中phenoData中的data
pdata <- pData(sCLLex)
group_list <- as.character(pdata[,2])
table(group_list)

3. 了解 str,head,help函数,作用于第二步提取到的表达矩阵。
# 展示内部结构
str(exprSet)
# 默认展示前6行
head(exprSet)
# 提供帮助文档
help("sCLLex")

4. 安装并了解hgu95av2.db包,看看ls(“package:hgu95av2.db”)后显示的那些变量。
# 使用bioconductor系列包的安装方法
BiocManager::install("hgu95av2.db")
suppressPackageStartupMessages(library( 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章
















 861
861











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









