






 本文详细介绍了单细胞转录组数据分析流程,包括实验平台选择、原始数据质控、文库拆分、细胞鉴定、定量分析等关键步骤,并探讨了不同标准化方法对表达数据的影响。
本文详细介绍了单细胞转录组数据分析流程,包括实验平台选择、原始数据质控、文库拆分、细胞鉴定、定量分析等关键步骤,并探讨了不同标准化方法对表达数据的影响。
单细胞系列教程
表达数据标准化理论
简介
上一步,我们鉴定出了重要的干扰因素和解释变量可能对表达定量带来影响。scater允许在后续统计模型中引入这些变量来屏蔽技术操作带来的影响,或者可以给函数normaliseExprs()提供一个设计矩阵design matrix来直接移除干扰因素的影响。在这一章先不涉及这些。
相反,我们探索下简单的量化因子size-factor标准化如何在校正文库大小的同时移除部分干扰因素引入的检测偏差。
文库大小
scRNA-seq数据的文库大小变化很大是因为混样测序,来源于每个细胞的总reads叔差别较大。一些定量方法,如Cufflinks, RSEM) 在估算基因表达时已经考虑了文库大小的影响因此不需要这一步标准化。
然而,如果采用的是其它的定量方法就必须首先通过某种方法估算一起比较的每个样品的文库大小也称为量化因子 (ormalization factor),然后原始表达量乘以或除以量化因子矩阵获得标准化后的表达结果。很多开发出来用于bulk RNA-seq的结果,也可以用于scRNA-seq,比如UQ, SF, CPM, RPKM, FPKM, TPM.
标准化方法
CPM
最简单的标准化方式是原始reads除以样品总的可用reads数乘以1,000,000获得每百万reads的count数 (counts per million(__CPM__)。因为考虑的是总的细胞内的reads,计算总的reads数时只考虑内源性基因而排除spike-ins部分的reads。CPM计算的R代码是:
calc_cpm <- function (expr_mat, spikes = NULL){
norm_factor <- colSums(expr_mat[-spikes,])
return(t(t(expr_mat)/norm_factor)) * 10^6
}这种计算方式的缺点是容易受到极高表达且在不同样品中存在差异表达的基因的影响;这些基因的打开或关闭会影响到细胞中总的分子数目,可能导致这些基因标准化之后就不存在表达差异了,而原本没有差异的基因标准化 之后却有差异了。
RPKM、FPKM和TPM是CPM按照基因或转录本长度归一化后的表达,也会受到这一影响。
为了解决这一问题,研究者提出了其它的标准化方法。
Relative Log Expression RLE (SF)
量化因子 (size factor, SF)是由DESeq [@Anders2010-jr]提出的。其方法是首先计算每个基因在所有样品中表达的几何平均值。每个细胞的量化因子(size factor)是所有基因与其在所有样品中的表达值的几何平均值的比值的中位数。由于几何平均值的使用,只有在所有样品中表达都不为0的基因才能用来计算,所以不适合大批量低深度的scRNA-seq数据。edgeR & scater 把这种方法称为”relative log expression” (RLE)。其在R中计算函数是:
calc_sf <- function (expr_mat, spikes=NULL){
geomeans <- exp(rowMeans(log(expr_mat[-spikes,])))
SF <- function(cnts){
median((cnts/geomeans)[(is.finite(geomeans) & geomeans >0)])
}
norm_factor <- apply(expr_mat[-spikes,],2,SF)
#return(t(t(expr_mat)/norm_factor))
return(norm_factor)
}上四分位数 (upperquartile, UQ)
上四分位数 (upperquartile, UQ)是样品中所有基因的表达除以该样品处于上四分位数的基因的表达值 [@Bullard2010-eb]。同时为了保证绝对表达水平的相对稳定,计算得到的上四分位数值要除以所有样品中上四分位数值的中位数。对低深度scRNA-seq数据,这个方法的一个缺点是可能处于上四分位数的基因的表达值为0或接近0。这个限制可以通过采用更高的分位数如99%分位数 (scater的默认值)或排除表达值为0的基因后剩余基因的上四分位数。示例如下:
calc_uq <- function (expr_mat, spikes=NULL){
UQ <- function(x) {
quantile(x[x>0],0.75)
}
uq <- unlist(apply(expr_mat[-spikes,],2,UQ))
norm_factor <- uq/median(uq)
#return(t(t(expr_mat)/norm_factor))
return(norm_factor)
}TMM (M-值的加权截尾均值)
TMM是M-值的加权截尾均值 [@Robinson2010-hz]。选定一个样品为参照,其它样品中基因的表达相对于参照样品中对应基因表达倍数的log2值定义为M-值。随后去除M-值中最高和最低的30%,剩下的M值计算加权平均值。每一个非参照样品的基 因表达值都乘以计算出的TMM。这个方法的两个可能问题是,一是Trim后没有足够的非0基因,另外该方法假设大部分基因的表达是没有差异的。
scran
scran采用为scRNA-seq设计的CPM方法的变种 [@LLun2016-pq]. 该方法通过把多组细胞合并到一起来屏蔽较多的0值问题,然后采用类似_CPM的方式计算标准化因子。因为一个细胞会出现在多个合并的集合里面 (pool),细胞特异的因子可以采用线性代数从非特异性因子中去卷积计算得来。(Since each cell is found in many different pools, cell-specific factors can be deconvoluted from the collection of pool-specific factors using linear algebra.)
Downsampling
最后一个校正文库大小的方式是对表达矩阵进行向下抽样使得每个细胞检测到的总分子数相同。这个方法的优势是计算过程中会引入0值进而消除不同细胞检测到的基因数不同引入的偏差。该方法最大的缺点是其非确定性,每次downsampling获得的表达矩阵都会有些细微不同。通常需要重复多次保证结果的稳定性。其操作代码是:
Down_Sample_Matrix <- function (expr_mat)
{
min_lib_size <- min(colSums(expr_mat))
down_sample <- function(x) {
prob <- min_lib_size/sum(x)
return(unlist(lapply(x, function(y) {
rbinom(1, y, prob)
})))
}
down_sampled_mat <- apply(expr_mat, 2, down_sample)
return(down_sampled_mat)
}标准化效果评估
我们将通过PCA方法和计算样品范围的 relative log expression (scater::plotRLE())来比较不同标准化方法的效果。含有更多reads的细胞,其大部分基因的表达比所有细胞的中值表达水平也更高,得到RLE值为正值;含有更少reads的细胞,其大部分基因的表达比所有细胞的中值表达水平更低,得到的RLE为负值。而标准化后的RLE值应该为0。计算_RLE_的函数如下:
calc_cell_RLE <- function (expr_mat, spikes = NULL)
{
RLE_gene <- function(x) {
if (median(unlist(x)) > 0) {
log((x + 1)/(median(unlist(x)) + 1))/log(2)
}
else {
rep(NA, times = length(x))
}
}
if (!is.null(spikes)) {
RLE_matrix <- t(apply(expr_mat[-spikes, ], 1, RLE_gene))
}
else {
RLE_matrix <- t(apply(expr_mat, 1, RLE_gene))
}
cell_RLE <- apply(RLE_matrix, 2, median, na.rm = T)
return(cell_RLE)
}注意 1: RLE, TMM, 和 UQ 量化因子方法是为bulk RNA-seq开发的,依赖于实验条件,不一定适合scRNA-seq数据,尤其是潜在假设不成立时。
注意 2: scater提供了一个函数calcNormFactors实现了几个文库大小标准化方法方便后续使用。
注意 3: edgeR对一些标准化方法做了额外的调整使得它可能获得与原始方法不同的结果。edgeR和scater的”RLE”方法基于DESeq的量化因子方法,但可能计算结果会不同于DESeq/DESeq2包的estimateSizeFactorsForMatrix计算的结果。另外,一些版本的edgeR只有在所有细胞的lib.size为设置为1时才计算标准化因子。
注意 4: CPM标准化使用的是scater包的calculateCPM()函数。scater包的normaliseExprs()函数用于 RLE, UQ 和 TMM 标准化计算。scran 标准化使用的是scran包计算量化因子 (基于SingleCellExperiment数据对象)和scater包的normalize()函数。所有标准化函数把计算结果存储到SCE对象的logcounts通道 (slot)。downsampling 标准化使用的是前面展示的方法。
标准化实战 (UMI)
继续使用tung数据集进行后续分析:
library(scRNA.seq.funcs)
library(scater)
library(scran)
options(stringsAsFactors = FALSE)
set.seed(1234567)
# umi <- readRDS("tung/umi.rds")
# umi.qc <- umi[rowData(umi)$use, colData(umi)$use]
# endog_genes <- !rowData(umi.qc)$is_feature_control
umi <- readRDS("tung/umi.rds")
umi_endog_genes <- !rowData(umi)$is_feature_control
umi_endog <- umi[umi_endog_genes,]
umi.qc <- umi[rowData(umi)$use, colData(umi)$use]
umi_qc_endog_genes <- !rowData(umi.qc)$is_feature_control
umi.qc_endog <- umi.qc[umi_qc_endog_genes,]原始值
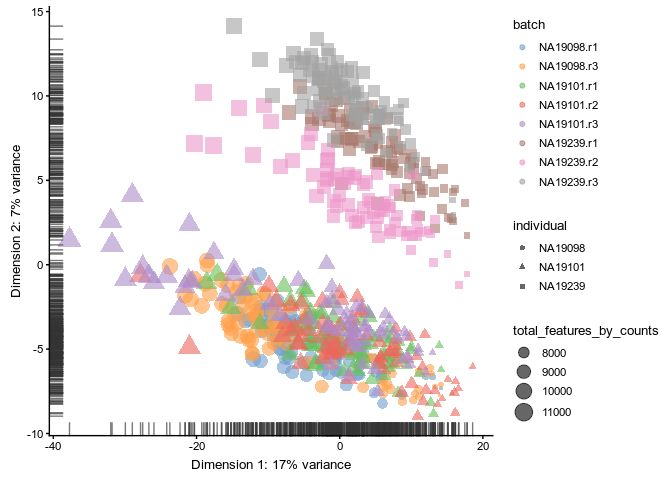
umi.qc_endog <- runPCA(umi.qc_endog, exprs_values = "logcounts_raw")
scater::plotPCA(
umi.qc_endog,
by_exprs_values = "logcounts_raw",
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
# plotPCA(
# umi.qc[endog_genes, ],
# exprs_values = "logcounts_raw",
# colour_by = "batch",
# size_by = "total_features",
# shape_by = "individual"
# )
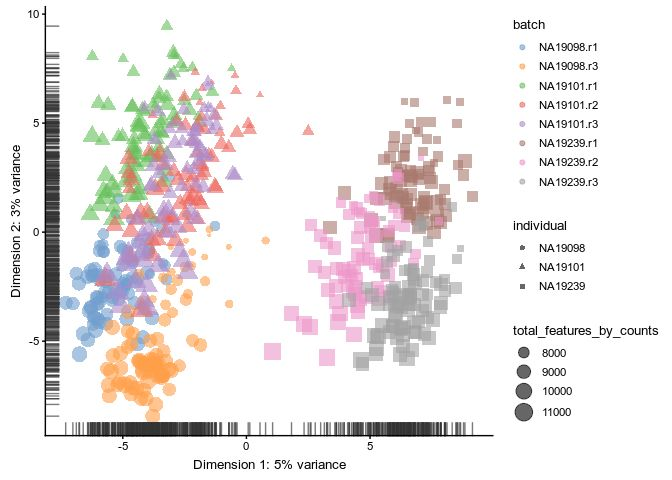
CPM
logcounts(umi.qc) <- log2(calculateCPM(umi.qc, use_size_factors = FALSE) + 1)
scater::plotPCA(
umi.qc[umi_qc_endog_genes,],
by_exprs_values = "logcounts",
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
library(cowplot)
# plotRLE(
# umi.qc[endog_genes,],
# exprs_mats = list(Raw = "logcounts_raw", CPM = "logcounts"),
# exprs_logged = c(TRUE, TRUE),
# colour_by = "batch"
# )
cpmlogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("Log CPM")
rawlogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts_raw",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("Raw log count")
plot_grid(rawlogcounts, cpmlogcounts, nrow=2, ncol=1)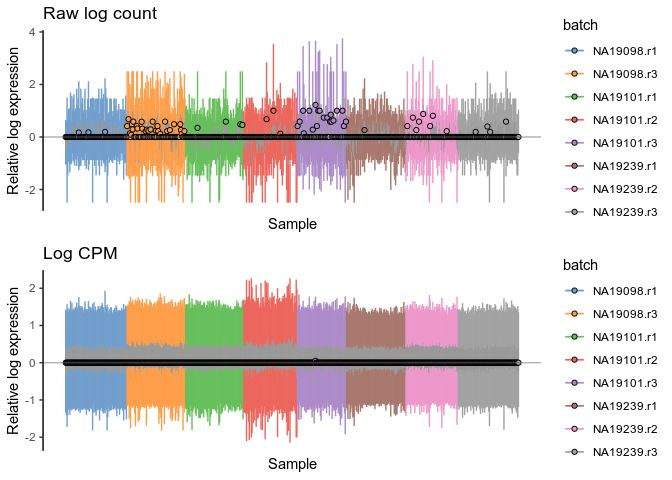
Size-factor (RLE)
# umi.qc <- normaliseExprs(
# umi.qc,
# method = "RLE",
# feature_set = umi_qc_endog_genes,
# return_log = TRUE,
# return_norm_as_exprs = TRUE
# )
sizeFactors(umi.qc) <- calc_sf(counts(umi.qc),
spikes=rowData(umi.qc)$is_feature_control_ERCC)
umi.qc <- normalize(umi.qc)
scater::plotPCA(
umi.qc[umi_qc_endog_genes,],
by_exprs_values = "logcounts",
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)rlelogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("RLE size factor")
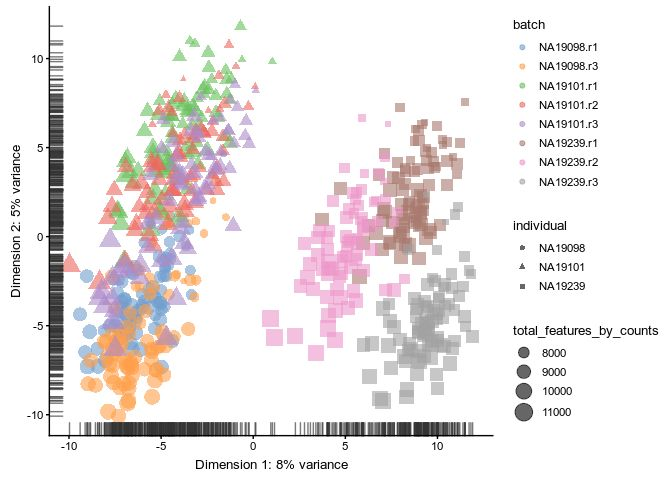
plot_grid(rawlogcounts, rlelogcounts, nrow=2, ncol=1)Upperquantile
# umi.qc <- normaliseExprs(
# umi.qc,
# method = "upperquartile",
# feature_set = endog_genes,
# p = 0.99,
# return_log = TRUE,
# return_norm_as_exprs = TRUE
# )
sizeFactors(umi.qc) <- calc_uq(counts(umi.qc),
spikes=rowData(umi.qc)$is_feature_control_ERCC)
umi.qc <- normalize(umi.qc)
plotPCA(
umi.qc[umi_qc_endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
uqlogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("UQ")
plot_grid(rawlogcounts, uqlogcounts, nrow=2, ncol=1)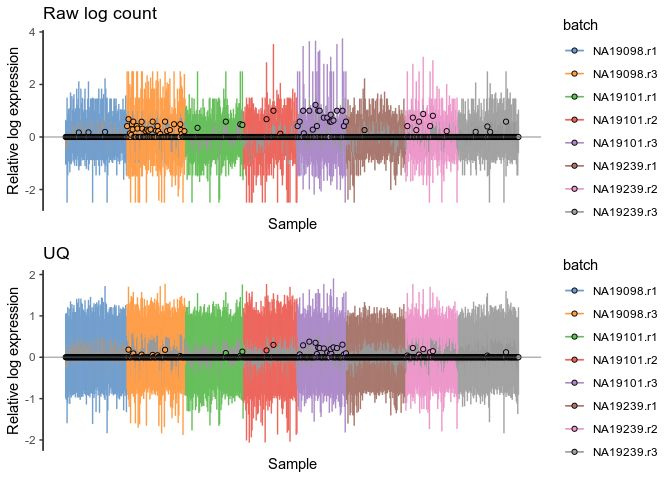
TMM
umi.qc <- normaliseExprs(
umi.qc,
method = "TMM",
feature_set = umi_qc_endog_genes,
return_log = TRUE,
return_norm_as_exprs = TRUE
)
plotPCA(
umi.qc[umi_qc_endog_genes, ],
colour_by = "batch",
size_by = "total_features",
shape_by = "individual"
)plotRLE(
umi.qc[umi_qc_endog_genes, ],
exprs_mats = list(Raw = "logcounts_raw", TMM = "logcounts"),
exprs_logged = c(TRUE, TRUE),
colour_by = "batch"
)scran
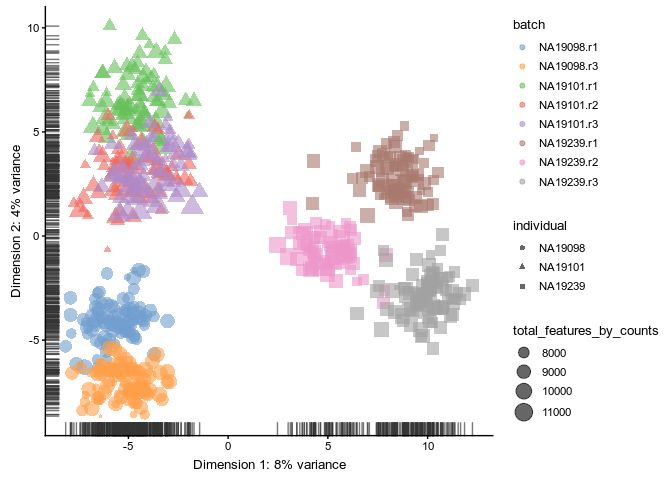
library(scran)
qclust <- quickCluster(umi.qc, min.size = 30)
umi.qc <- computeSumFactors(umi.qc, sizes = 15, clusters = qclust)
umi.qc <- normalize(umi.qc)
plotPCA(
umi.qc[umi_qc_endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
scranlogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("Scran")
plot_grid(rawlogcounts, scranlogcounts, nrow=2, ncol=1)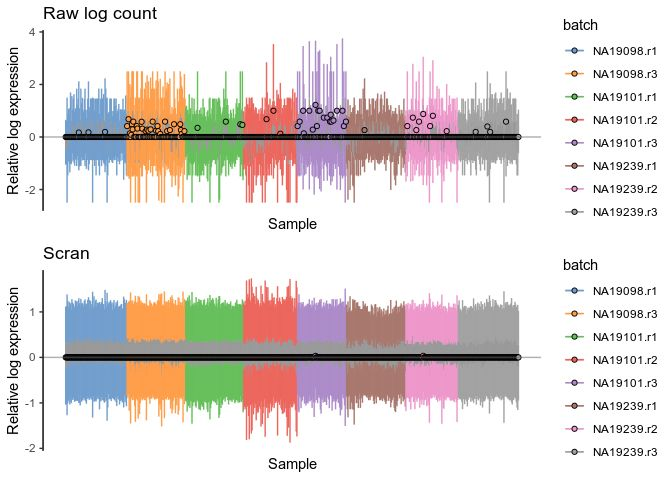
scran有时会获得负或零量化因子,这将会严重干扰标准化后的表达矩阵,需要采用下面的方法确认没有问题:
summary(sizeFactors(umi.qc))## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.4671 0.7787 0.9483 1.0000 1.1525 3.1910对这个数据集来说,量化因子是合理。如果计算时发现scran给出的量化因子是非正值尝试增加cluster和pool的大小,直到获取正值。
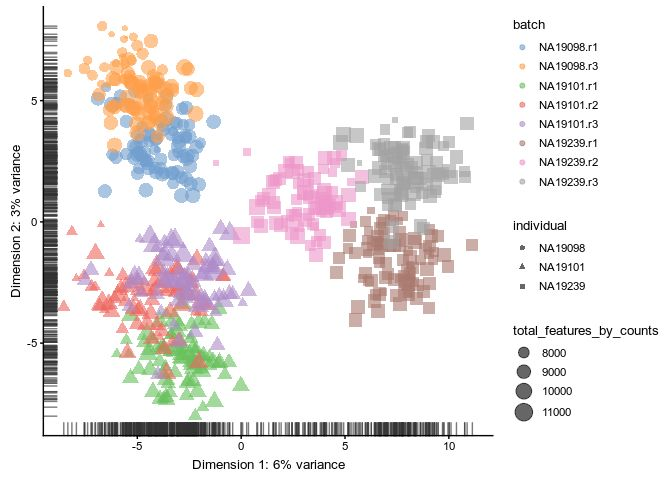
Downsampling
logcounts(umi.qc) <- log2(Down_Sample_Matrix(counts(umi.qc)) + 1)
plotPCA(
umi.qc[umi_qc_endog_genes, ],
colour_by = "batch",
size_by = "total_features_by_counts",
shape_by = "individual"
)
dslogcounts <- plotRLE(
umi.qc[umi_qc_endog_genes,],
exprs_values = "logcounts",
exprs_logged = c(TRUE),
colour_by = "batch"
) + ggtitle("Downsampling")
plot_grid(rawlogcounts, dslogcounts, nrow=2, ncol=1)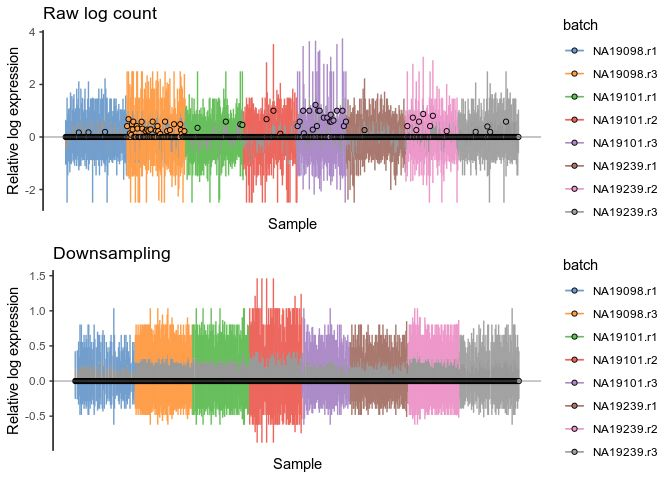
易生信系列培训课程,扫码获取免费资料

更多阅读
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











