







时间:2021-11-15
单位:英国NIHR生物医学研究中心
期刊:Journal of Medical Genetics (JMG, IF=6.311)
题目:多变的骨骼表型与PRKG2双等位基因变异相关
链接:http://dx.doi.org/10.1136/jmedgenet-2021-108027
前 言
十万基因组计划 (100KGP)是一项英国的国家倡议,目标是使用全基因组测序 (WGS)来识别罕见遗传疾病的致病原因,并将这项技术的使用嵌入到国民医疗保健 (National Health Service, NHS)制度中。利用这一资源的数据以及国际基因匹配工作 (International gene-matching efforts),来自两个独立家系的4个人被鉴定出在PRKG2 (一种最近描述的骨骼发育不良基因)中存在纯合移码变异或终止 (Stop-gain)变异。
PRKG2编码环状鸟苷依赖蛋白激酶II (Cyclic guanosine monophosphate dependent protein kinase II, cGKII),位于NPR-B/CNP下游。NPR-B由NPR2编码,其双等位基因变异导致Maroteaus型肢端发育不全 (Acromesomelic dysplasia, Maroteaus type, AMDM) (MIM编号 602875)。啮齿动物模型也表明PRKG2与骨骼发育有关,并且cGKII缺乏症是造成安格斯牛 (Angus cattle)的侏儒症表型的原因。
在这项研究中,作者通过英格兰基因组学公司提供的LabKey应用程序,使用来自100KGP的数据搜索了罕见的PRKG2双等位基因变异。研究人员可以在线申请访问 (www.genomicsengland.co.uk/join-a-gecip-domain)。最初的筛选基于1000G数据库和内部人群频率信息,采用了1%的等位基因人群频率阈值,并使用ACMG标准对变异进行了分类。
结 果
在家族1中,通过WGS和Sanger测序在3个患有椎体-骺端发育不良 (Spondylometaphyseal dysplasia, SMD)的兄弟中发现了一个纯合子致病突变PRKG2 (NM_006259.3:c.2282dup, p.Asp761Glufs*34)。有趣的是,F1-IV-6患者 (中度致病)也有I型成骨不全 (Type I osteogenesis imperfect, OI)。一项早期临床外显子组测序研究发现,在分子诊断的病例中,有4.6%的病例有一个以上的基因变异导致混杂 (Blended)表型。
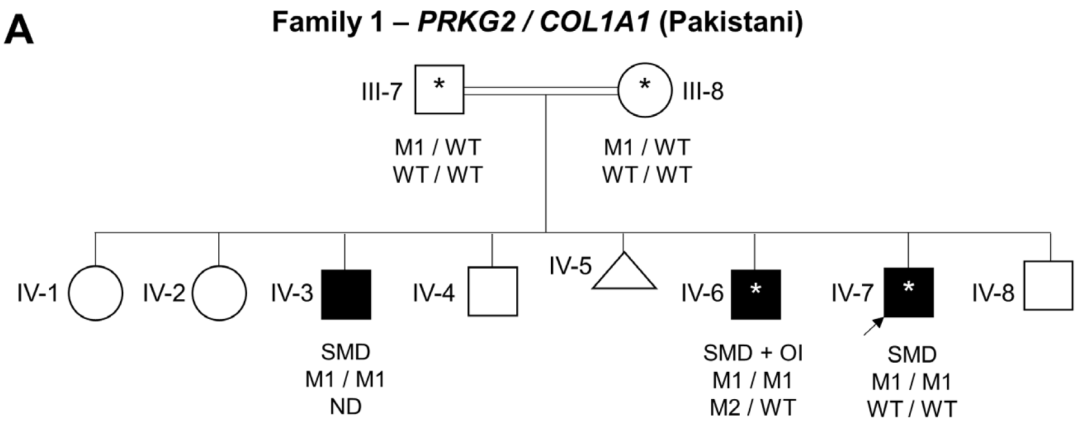
家系1家系分析图(双线为近亲婚配),箭头先证者M1/M1突变

这样的复杂病例预计在近亲的家庭中更为常见,大范围纯合子区域 (ROHs)占基因组的很大比例;然而,对于F1-IV-6,OI的二次诊断是由于COL1A1移码引起的,这是从头开始出现的。这名儿童因儿童时期多发骨折 (婴儿时手臂、8岁手腕和胸部T6楔形骨折)伴有蓝色巩膜而怀疑患有OI。当然,同时存在的OI可能对这个个体的表型严重程度有影响,尤其是因为他的身高比他的两个兄弟更低,OI (1型)是一种已知的导致身材矮小的原因。
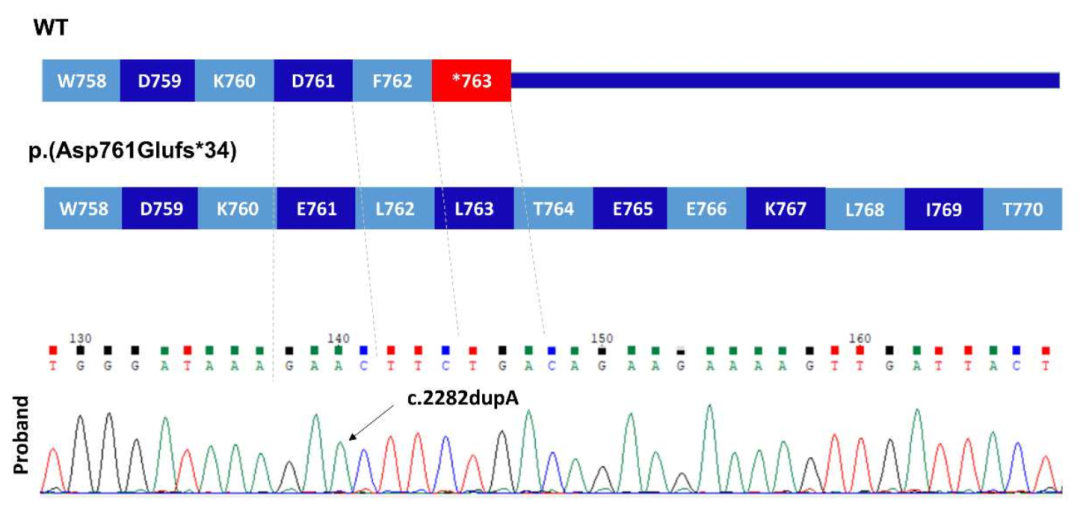
家系1一代测序验证
插入A碱基后发生移码,且无法终止翻译
在家系2中,一个患有肢端发育不良的女孩的外显子组测序显示了一个纯合子致病变异PRKG2c.1705C>T;(Arg569*)。将F2-V-3的基因组数据与之前发表的病例进行比较,未能检测到PRKG2基因座 (Locus)上共有的单倍型。c.1705 C>T的复发似乎更有可能是由于单独的突变事件。
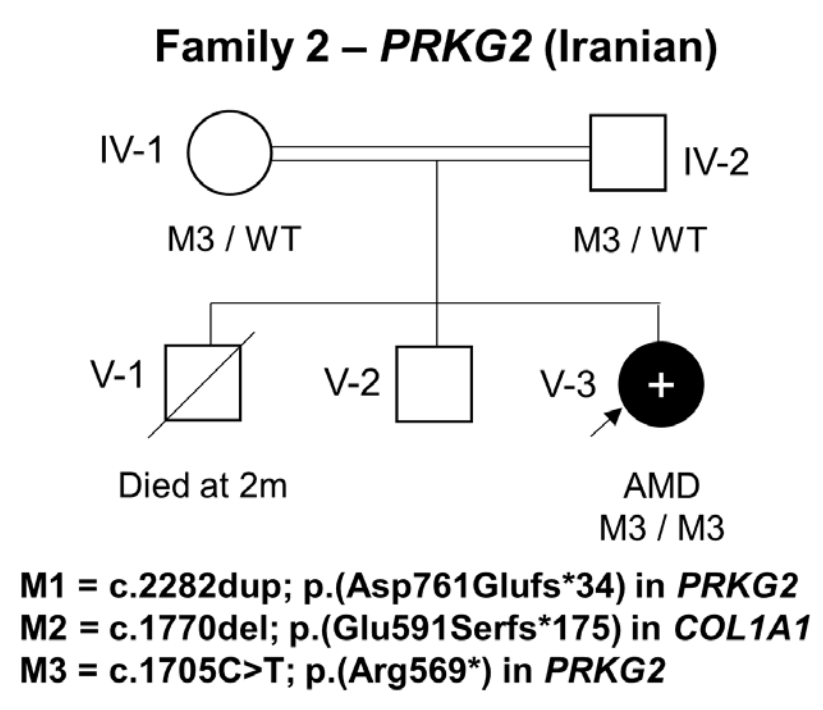
家系2家系分析图,女孩M3/M3突变
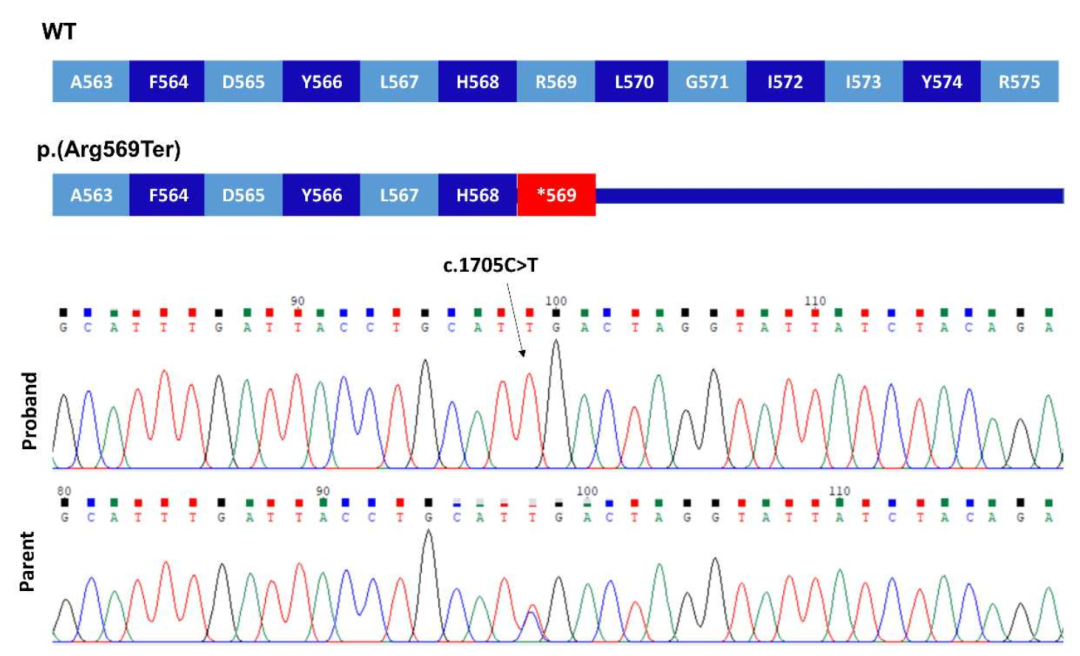
家系2一代测序验证,突变后翻译异常终止
这里描述的这两种变异都非常罕见,gnomAD数据库中没有p.Asp761Glufs*34,而p.Arg569*为单基因。在这两个家系中,致病变异都存在于较大的ROHs中。虽然p.Arg569*突变已经被证明影响下游的MAPK途径,但家系1中的p.Asp761Glufs*34很可能也是有害的,因为蛋白C末端Asp>Phe及33个氨基酸发生替换。在计算机建模中的研究突出了该区域的结构重要性,特别是终末Phe762残基。
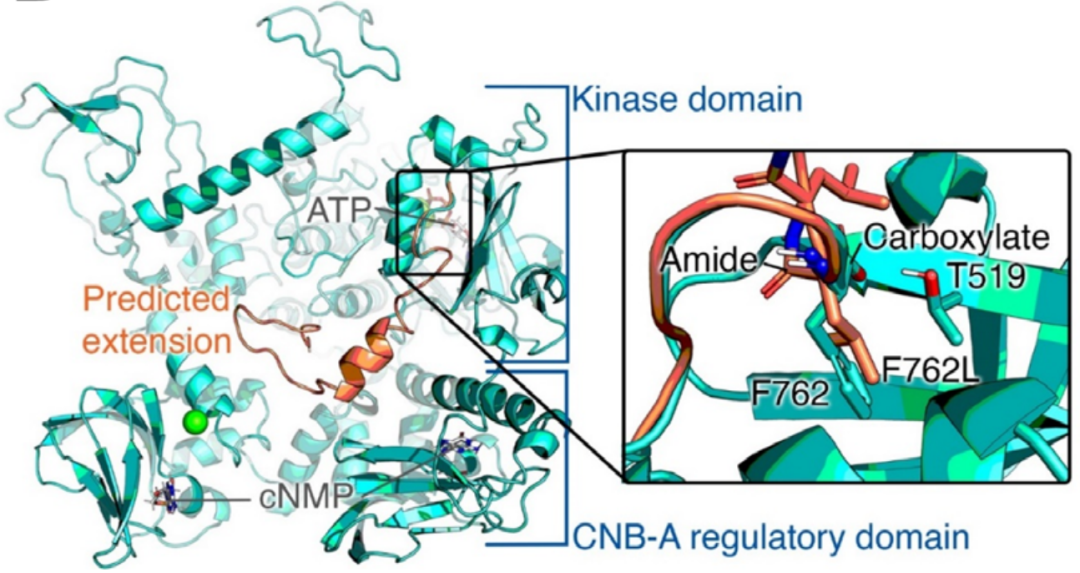
cGKII (野生型: 绿松石色)的结构,与突变体 (橙色,762突变且多出一段)
为了从功能上证实新发现的p.Asp761Glufs*34变异的致病性,我们首先用Western blot分析了cGKII的表达。以先前鉴定的p.Arg569*突变作为阳性对照,在这两个突变中,cGKII被检测到的大小与预测的大小一致,尽管与野生型相比,cGKII的水平显著降低 (≥80%)。接下来,我们通过分析p.Asp761Glufs*34突变体诱导Raf-1在Ser-43和ERK1/2磷酸化的能力,评估了它是否能够抑制FGF2诱导的MAPK通路。结果显示,p.Asp761Glufs*34突变体未能在Ser-43处磷酸化Raf-1,因此降低了FGF2诱导的ERK1/2磷酸化,这与p.Arg569*变体的结果相似。
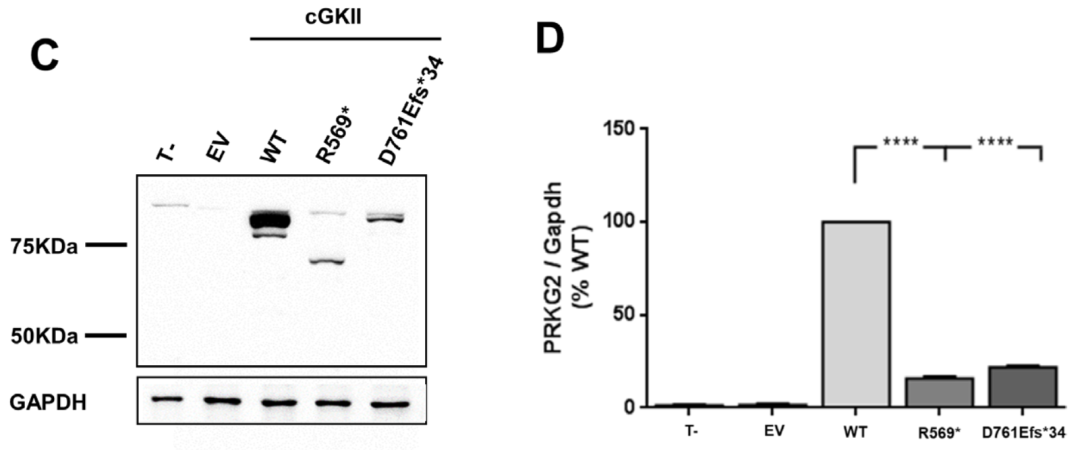
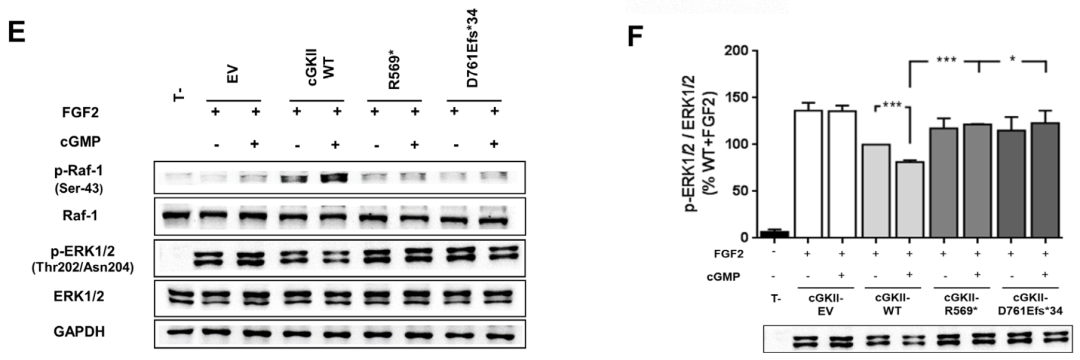
PRKG2变异对cGKII蛋白水平/MAPK通路调控的影响
本研究提供了两个家系的详细表型信息,并与两个已发表的病例进行了比较。F2-V-3的放射学发现与之前描述的“先证者1”的放射学发现非常相似,这并不令人惊讶,因为这两个人都有相同的纯合子p.Arg569*。相反,PRKG2家族1有一致的放射学表型,不同于先前报道的AMDP和AMDM。
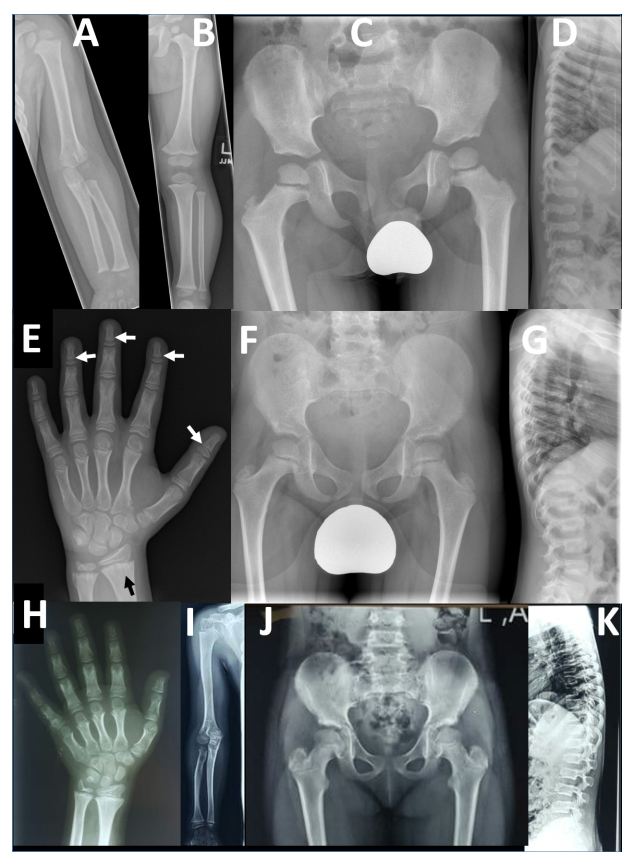
两个PRKG2变异家族的影像学表现
本文报告的三兄弟 (F1-IV-3、IV-6和IV-7)除了个别F1-IV-7有轻微的脚趾缩短外,没有肢端缩短 (Acromesomelic shortening)的证据。主要发现是颈椎病伴有椎体前突、股骨颈细长、干骺端不规则 (最明显的是桡骨和尺骨)和横纹(Striations)。其中一个儿童的远端指骨干骺端呈圆锥形,但是不显著。综上所述,家族1表现为以脊柱干骺端发育不良 (Spondylometaphyseal dysplasia)为特征的骨骼表型,而不是像AMDP和AMDM所预期的肢端发育不全。
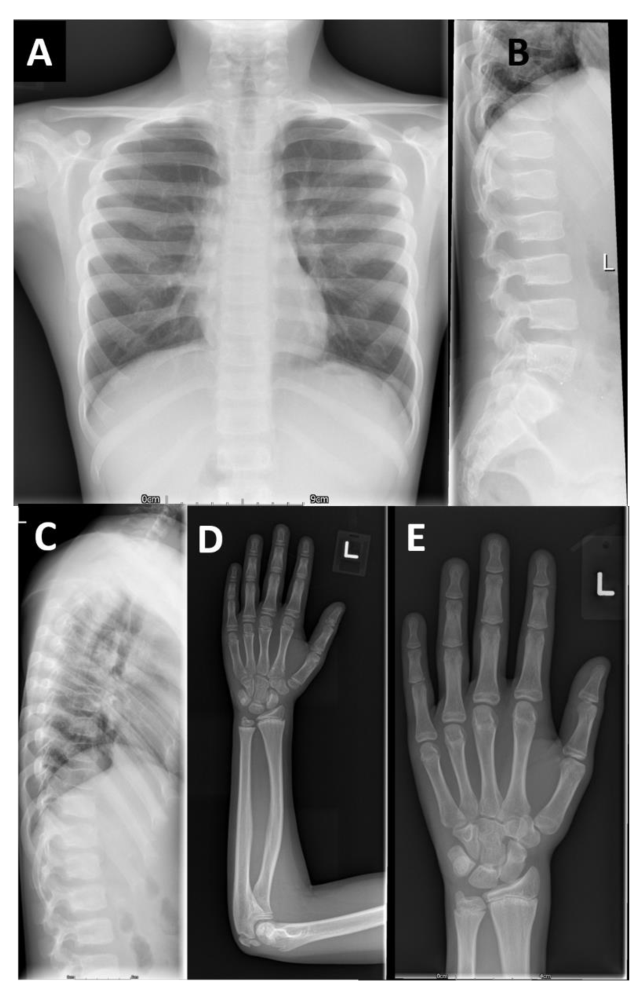
F1- IV- 7的额外影像学表现
结 论
综上所述,本研究利用对100KGP数据的分析,确定了发生PRKG2基因功能变异 (双等位基因缺失)的4个受影响个体。这些病例的表型范围包括脊柱干骺端发育不良等。这里描述的患者是在100KGP范围内所有罕见疾病中仅有的具有严重的PRKG2双等位基因变异的个体,且在人类中极其罕见。
欢迎咨询全固态大型云服务器租用
1周内完成家系变异分析,尽快推进下游分析
更适合家系全外显子组

若有服务器亦可免费技术咨询,提供力所能及的解答
一/二代测序、临床基因组/外显子组/转录组、遗传学分析


第二期临床基因组家系数据分析实战,快速发表SCI文章
系统性培训,一次学会终身会分析,只待新病例
服务器免费1个月,每日答疑,足以完成小家系分析
— 基本概念 —
— 文献解读 —
基因突变与脑瘫风险(Nature Genetic,2020)
— 数据库 —
ClinVar数据库详解
在线人类孟德尔遗传 (OMIM)数据库简介
— 期刊 —
— 分析技术 —
Jalview多序列比对图中显示序列标识
— 分析平台 —
Linux操作系统结构及常用命令
设置RStudio-Server不频繁掉线
RStudio-Server安装和内网穿透要点
Linux服务器的磁盘概念与相关操作 (一)
Linux服务器的磁盘概念与相关操作 (二)
— 政策法规 —
雇人代写论文是否犯法?
中华人民共和国人类遗传资源管理条例
— Tales of Genetics —
高中学历父亲自学基因编辑,看五六百篇论文,自制药用级化合物救治罕见病儿子!
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集





















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











