









 本文介绍了SAM和BAM文件格式,这两种格式广泛应用于存储由下一代测序技术产生的核酸序列数据。SAM是一种纯文本格式,而BAM是其二进制压缩版本。文章详细解析了SAM文件的组成结构,包括header部分和本体部分的数据记录方式,以及如何通过多种工具如samtools、hitslib等进行文件处理。
本文介绍了SAM和BAM文件格式,这两种格式广泛应用于存储由下一代测序技术产生的核酸序列数据。SAM是一种纯文本格式,而BAM是其二进制压缩版本。文章详细解析了SAM文件的组成结构,包括header部分和本体部分的数据记录方式,以及如何通过多种工具如samtools、hitslib等进行文件处理。
Sam文件
SAM file format wiki :
Sequence Alignment Map (SAM) is a text-based format originally for storing biological sequences aligned to a reference sequence developed by Heng Li and Bob Handsaker et al. It is widely used for storing data, such as nucleotide sequences, generated by next generation sequencing technologies, and the standard has been broadened to include unmapped sequences
-
sam 文件是国际顶级生物信息学专家李恒博士在冷泉港捣鼓出来的一种文件格式,储存了侧获得的信息,map到基因组后的各种信息。sam文件为纯文本格式,信息量极大。
-
BAM则是SAM的二进制格式。在sam的基础上应用上二进制编码,极大压缩了sam文件的体积。
常用的alignment的软件大都支持SAM/BAM格式,围绕这个文件李博士还开发了多个软件和多个不同语言的依赖库。以便大家使用该文本格式储存信息,并在改文件基础上进行开发。
- samtools:对SAM/BAM等格式进行各种相关操作的软件,功能最丰富。
- hitslib:samtools的C接口,可以通过该库,在C语言中完成samtools的同等功能。
- hitsjdk:java接口。
- pysam:python接口。
具体使用方法请查看相关文档
格式
以sam格式为例
@HD VN:1.6 SO:coordinate
@SQ SN:ref LN:45
r001 99 ref 7 30 8M2I4M1D3M = 37 39 TTAGATAAAGGATACTG *
r002 0 ref 9 30 3S6M1P1I4M * 0 0 AAAAGATAAGGATA *
r003 0 ref 9 30 5S6M * 0 0 GCCTAAGCTAA * SA:Z:ref,29,-,6H5M,17,0;
r004 0 ref 16 30 6M14N5M * 0 0 ATAGCTTCAGC *
r003 2064 ref 29 17 6H5M * 0 0 TAGGC * SA:Z:ref,9,+,5S6M,30,1;
r001 147 ref 37 30 9M = 7 -39 CAGCGGCAT * NM:i:1
SAM文件主要由两个部分构成
-
header:标记了该SAM文件的一些基本信息,比如版本、按照什么方式排序的、Reference信息等等。
-
本体:每行为一个reads,不同列记录了不同的信息,列与列之间通过tab分隔。
格式的意义
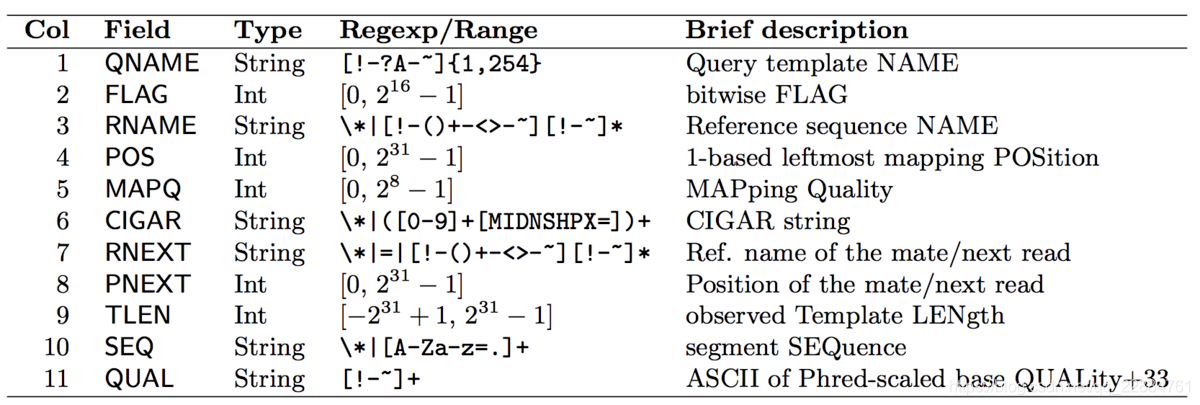
- QNAME:测序的reads的名字。
- FLAG:二进制数字之和,不同数字代表了不同的意义;比如正负链,R1/R2(双端 测序的哪一端)等。
- RNAME:map到参考基因组后的染色体名称。
- POS:1-based 基因组起始位点。
- MAPQ:map的质量。
- CIGAR:一个数字与字母交替构成的字符串,标记了这段reads不同位置的match情况。不同字母的含义后边介绍。
- RNEXT:如果是pair-end测序,这个为mate(另一端中对应的)的read的染色体名称;否则为下一条read的染色体名称。
- PNEXT:同上,read对应的起始位点。
- TLEN:模板的长度。
- SEQ:序列。
- QUAL:序列的质量打分(fasta文件中的那个)。
flag的含义

flag比较特殊的是一点在于flag最后的数字中包含了这个数即为true(包含该特性),不包含这个数字即为false(不包含该特性)。
检测方法也比较巧妙,通过二进制与进行计算。比如:16 and 16 -> 16。结果与测试的数字一致,则表明flag中包含该数字。否则,不包含。
当然,不同语言版本的接口也直接提供了api来直接获取这些特性,不必在进行人工的计算。
cigar的含义
cigar中会包含数字,代表了特定match持续了多少nt;以及不同的字符,代表了不同的match情况。
30S512M216N12S (30nt soft clip -> 512nt exact match -> 216nt skipped region -> 12nt soft clip)
30S (30nt soft clip)
40M (40nt exact match)
其中不同的字符及其含义如下:

注意 双端测序,正负链的问题
双端测序很麻烦,mate和read两条序列的flag信息基本都是完全相反的,包括strand等信息。那么如何判断这一对reads测到的到底是哪条链就成了问题。
- 最稳妥的方法是,通过GT/AG规则来确定。缺点就是你很难提取到GT/AG这个序列,最起码在我的测试中如此。
- 在我的理解中,cigar中的I、D、N三个字符代表的区域不计入位点序列中。那么N区域的第一个位点周边的序列即为内含子周边的序列,然而在这个位点周边,很难获取到标准的GT/AG序列及其互补序列。可能由junctions的突变造成的,即N区域并不一定是标注的内含子区域。
- STAR应该是通过这个途径识别链方向的 。
alignSJstitchMismatchNmax 0 -1 0 0
4*int>=0: maximum number of mismatches for stitching of the splice junctions (-1: no limit).
(1) non-canonical motifs, (2) GT/AG and CT/AC motif, (3) GC/AG and CT/GC motif, (4) AT/AC and GT/AT motif.
- 另外就是根据readNegativeStrand和mateNegativeStrand,配合以First in pair以及second in pair进行判断。
- 这个办法,依赖于是否确定R1和R2的建库方式和建库方向,如果我们确定R1是某个特定方向,那么我们就能够通过这四个参数的组合获取到正确的方向。
- 目前我已知的regtools就是通过这种途径进行链的识别的。
//Get the strand from the bitwise flag
void JunctionsExtractor::set_junction_strand_flag(bam1_t *aln, Junction& j1) {
uint32_t flag = (aln->core).flag;
// 从flag中提取出readNegativeReversed,mateNegativeReversed, first in pair和second in pair的信息
int reversed = (flag >> 4) % 2;
int mate_reversed = (flag >> 5) % 2;
int first_in_pair = (flag >> 6) % 2;
int second_in_pair = (flag >> 7) % 2;
// 下面就是判断正负链的过程
// strandness_ is 0 for unstranded, 1 for RF, and 2 for FR
int bool_strandness = strandness_ - 1;
int first_strand = !bool_strandness ^ first_in_pair ^ reversed;
int second_strand = !bool_strandness ^ second_in_pair ^ mate_reversed;
char strand;
if (first_strand){
strand = '+';
} else {
strand = '-';
}
//cerr << "flag is " << flag << endl;
// if strand inferences from first and second in pair don't agree, we've got a problem
if (first_strand == second_strand){
j1.strand = string(1, strand);
} else {
j1.strand = string(1, '?');
}
//cerr <<"flag strand is " << j1.strand << endl;
return;
}
- 根据参数推测,hisat2可能也是这个方式。
--fr/--rf/--ff -1, -2 mates align fw/rev, rev/fw, fw/fw (--fr)


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









