







这篇文章主要写介绍16S测序的相关知识,之后如果我有时间会写16S的数据处理,设计生物信息学的内容。
宏基因组学
宏基因组( M e t a g e n o m e Metagenome Metagenome),也称微生物环境基因组或元基因组。是由Handelsman等1998年提出的新名词,其定义为"the genomes of the total microbiota found in nature",即生境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因,目前主要指环境样品中的细菌和真菌的基因组总和。
宏基因组学研究对象是特定环境中的总DNA,不是某特定的微生物或其细胞中的总DNA,不需要对微生物进行分离培养和纯化,这对我们认识和利用95%以上的未培养微生物提供了一条新的途径。
扩增子测序
扩增子是DNA或RNA扩增后的一段核苷酸序列。比如通过PCR扩增得到的某个基因的扩增片段。扩增子测序是对特定长度的PCR产物或者捕获的片段进行测序,分析序列中的变异。扩增子测序是一种高靶向性方法,用于分析特定基因组区域中的基因变异。扩增子测序能够根据不同需求,对目标区域进行高覆盖度的测序,还可以检测到低频突变。
扩增子测序主要包括16S rDNA测序、18S rDNA测序、ITS测序及目标区域扩增子测序等。 16S/18S/ITS rDNA,序列中都包含可变区(Variable region)和保守区(constant region)。保守序列区域反映了生物物种间的亲缘关系,而可变序列区域则能体现物种间的差异。可变区序列因细菌而异,且变异程度与细菌的系统发育密切相关,通过提取环境样品的DNA,并扩增和检测其中的一段或几段区域,可以了解环境样品中细菌的分类和丰度(丰度:群落内物种数目的多少)。
全基因组测序
1、当前的全基因组测序费用较高,单存用于物种鉴定并不划算;
2、全基因组测序的数据,用于分析需要消耗非常多的计算资源,以及人力物力;
3、全基因组测序数据量大,比对需要消耗更多的时间,这对于一些微生物的快速检测显然是不利的;
4、目前微生物全基因组的数据库资源不够丰富,很多序列依然无法比对到数据库中;
微生物
定义:个体难以用肉眼观察的一切微小生物之统称。
微生物包括细菌、病毒、真菌和少数藻类等。(但有些微生物是肉眼可以看见的,像属于真菌的蘑菇、灵芝等。所以定义里难以是不容易看见,不是看不见嗷,别较真)。
根据存在的不同环境分为空间微生物、海洋微生物等,按照细胞结构分类分为原核微生物和真核微生物。
它个体微小,与人类关系密切。涵盖了有益跟有害的众多种类,广泛涉及食品、医药、工农业、环保、体育等诸多领域。
微生物鉴定
这些都是了解即可,毕竟讲的是16S,这个主要是说这些方法都有局限性,对比衬托16S嘛(•͈ᴗ•͈ૢૢ)❊⿻*
形态学
形态结构特征:形态学特征始终被用作微生物分类和鉴定的重要依据之一,其中有两个重要原因:
- 它易于观察和比较,尤其是在真核微生物和具有特殊形态结构的细菌中;
- 许多形态学特征依赖于多基因的表达,具有相对的稳定性。常用于原核生物分类鉴定的形态学特征。
常用微生物分类鉴定的形态学特征
- 培养特:最重要是菌落特征:如菌落的形状、大小、颜色、隆起、表面状况、质地、光泽及水溶性色素等
- 细胞形态
- 形状:球形、杆状、弧形、螺旋形、丝状、分枝及特殊形状
- 大小:其中最重要的是细胞的宽度或直径
- 排列:单个、成对、成链或其他特殊列方式
- 特殊的细胞结构
- 鞭毛:有无鞭毛、着生位置及其数量
- 芽孢:有无芽孢、形状、着生位置及孢囊是否影大
- 孢子:形状、着生位置、数量及排列
- 其他:类膜、细胞附属物,如柄、丝状物、粘、蓝细菌的异形跑、静止细胞和连锁体等
- 超微结构:细胞壁、细胞内膜系统、放线菌孢子表面特征等
- 细胞内含物:异染颗粒、聚β-羟丁酸等类脂颗粒、硫粒、气泡、伴孢品体等
- 染色反应:革兰氏染色、抗酸性染色
- 运动性:鞭毛泳动、常行、螺旋体运动方式
生理学
生理生化特征
生理生化特征和微生物的酶和调节蛋白质的本质和活性直接相关酶及蛋白质都是基因的产物。对微生物生理生化特征和比较也是对微生物基因组的间接比较,加上测定生理生化特征比直接分析基因组要容易得多。因此生理生化特征对于微生物的系统分类仍然是有意义的。
常用于微生物分类鉴定的生理生化特征
- 营养类型:光能自养、光能异养、化能自养、化能异养及兼性营养型
- 对氮源的利用能力:对蛋白质、蛋白胨、氨基酸、含氮无机盐及N:等的利用
- 对碳源的利用能力:对各种单糖、双糖、多糖、醇类及有机酸等的利用
- 对生长因子的需要:对特殊维生素、氨基酸、X-因子及V-因子等的依赖性
需氧性 - 对温度的适应性:最适、最低及最高生长温度和致死温度
- 对pH的适应性:在一定pH条件下的生长能力及生长的pH范固
- 对渗透压的适应性:对盐浓度的耐受性或嗜盐性
- 对抗生素及抑菌剂的敏感性:对抗生素、氰化钾(钠)、胆汁、弧菌抑制剂或某些染料的敏感性
- 产生某些酶类:如过氧化氨酶、氧化酶、酯酵、尿酶及DNA酶等
- 代谢产物:各种特征性代谢产物
- 与宿主的关系:共生、寄生、致病性等
噬菌体
噬菌体分型
在原核生物中已普遍发现有相应种类的噬菌体。噬菌体对宿主的感染和裂解作用具有高度的特异性,即一种噬菌体往往只能感染或裂解某种细菌,甚至只裂解种内的某些菌株。所以,根据噬菌体的宿主范围可将细菌分为不同的噬菌型和利用噬菌体裂解这样的特异性进行细菌鉴定。
血清型
血清学试验
细菌细胞与病毒等都含有蛋白质、脂白蛋、脂多糖等有抗原性的物质,由于不同微生物抗原物质结构不同,赋予它们不同的抗原特征。一种细菌的抗原除了可与它自身的抗体起特异性免疫反应外,若它与其它种类的细菌有共同的抗原组分,它们的抗原抗体之间就会发生交叉反应。
核酸碱基组成
核酸的碱基组成
分类学上,用
G
+
C
G+C
G+C占全部碱基的物质的量百分数
(
G
+
C
)
%
(G+C)\%
(G+C)%来表示各种生物的DNA碱基组成特征。
每一种生物都有一定的碱基组成亲缘关系近的生物,它们应该具有相似的G+C含量,若不同的生物之间
G
+
C
G+C
G+C含量差别大表明它们关系远,现有数据表明:
- 高等植物的 G + C G+C G+C含量范围大约 35 % − 50 % 35\% - 50\% 35%−50%之间;
- 原核生物中 G + C G+C G+C含量变化幅度宽达 22 % − 80 % 22\% - 80\% 22%−80%;
- 脊椎动物 G + C G+C G+C含量约 35 % − 45 % 35\% - 45\% 35%−45%之间;
由于原核生物中
G
+
C
G+C
G+C含量变化幅度宽达
22
%
−
80
%
22\% - 80\%
22%−80%;,这也足以表明原核生物是一个极其多样性的类群。
同一个种内的不同菌株
G
+
C
G+C
G+C含量差别应在
4
%
−
5
%
4\%-5\%
4%−5%以下;同属不同种的差别在
10
%
−
15
%
10\%-15\%
10%−15%,通常低于
10
%
10\%
10%。
分子杂交
生物的遗传信息以(遗传密码)形式线性的排列在DNA分子中,不同DNA碱基排列顺序的异同直接反映这些生物之间亲缘关系的远近,碱基排列顺序差异越小,它们之间的亲缘关系越近,反之亦然。
- DNA-DNA杂交:亲缘关系相对近的微生物之间的亲缘比较
- DNA-RNA杂交:亲缘关系相对远的微生物之间的亲缘比较
- 核酸探针:利用特异性的探针,用于细菌等的快速鉴定
16S测序
通过16S测序,我们可以知道一个样本中包含了哪些微生物,微生物的丰度如何,不同样品微生物的组成和丰度有哪些差别,同一样品不同外界条件下微生物的组成和丰度有哪些差别。
S-沉降系数
S为沉降系数,当用超速离心法测定一个粒子的沉淀速度时,此速度与粒子的大小直径成比例。5S即含有120个核苷酸,16S含有1540个核苷酸,而23S含有2900个核苷酸。
超速离心
- 差速离心:交替使用低速和高速离心,用不同强度的离心力使具有不同质量的物质分级分离的方法。此法适用于混合样品中各沉降系数差别较大组分的分离。
- 密度梯度离心:分离沉降系数接近的物质。使用一种密度能形成梯度(在离心管中,其密度从上到下连续增高)又不会使所分离的生物活性物质凝聚或失活的溶剂系统,离心后各物质颗粒能按其各自的比重平衡在相应的溶剂密度中形成区带。
常用的密度梯度溶剂是蔗糖或氯化铯(CsCl)溶液。用蔗糖时,先将蔗糖溶液制成密度梯度溶液,再在其顶端加样品。离心后,如欲收集所分离的组分,可在离心管的下端刺一小洞,然后分部收集。如用CsCl这种密度大又扩散迅速的溶剂系统时,可将样品均匀地混合于溶剂中。离心达到平衡后, CsCl溶液形成密度梯度,样品中各组分也在相应密度处形成区带。
16SrDNA和16SrRNA
- 16S rRNA:这并不是我们的研究对象,因为我们测序的不是它,而是它对应在DNA双链上的基因序列,
- 16S rDNA:我们所说的16S 就是指 16S rDNA。
rRNA
又称核糖体RNA( r i b o s o m a l R N A ribosomal RNA ribosomalRNA),rRNA是组成核糖体的主要成分。核糖体是合成蛋白质的工厂。rRNA一般与核糖体蛋白质结合在一起,形成核糖体( r i b o s o m e ribosome ribosome)。
rDNA
是细菌染色体上编码rRNA相对应的DNA序列,存在于所有细菌染色体基因中。
二者关系
rDNA指的是基因组中编码rRNA(核糖体RNA)分子的对应的DNA序列,也就是编码rRNA的基因。 rRNA是rDNA的转录产物,它是构成核糖体的重要成分,核糖体由许多小的rRNA分子组装而成。16S rRNA为核糖体RNA的一个亚基,16S rDNA就是编码该亚基的基因。一般所分析的对象都是16s rDNA。
为什么选用16S rDNA
随着生物技术的飞速发展,传统的微生物鉴定方法常常难以鉴定众多的生长习性复杂的微生物,因而基于基因组序列的分子鉴定受到广泛关注。
细菌rRNA(核糖体RNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列,存在于所有细菌染色体基因中。16S rDNA是细菌的系统分类研究中最有用的和最常用的分子钟:
分子钟: 随着不同生物来源的大量蛋白质序列的确定,Zucherkandl等发现:某一蛋白在不同物种间的取代数与所研究物种间的分歧时间接近正线性关系(即氨基酸在单位时间以同样的速度进行置换),进而将分子水平的这种恒速变异称为“分子钟”。
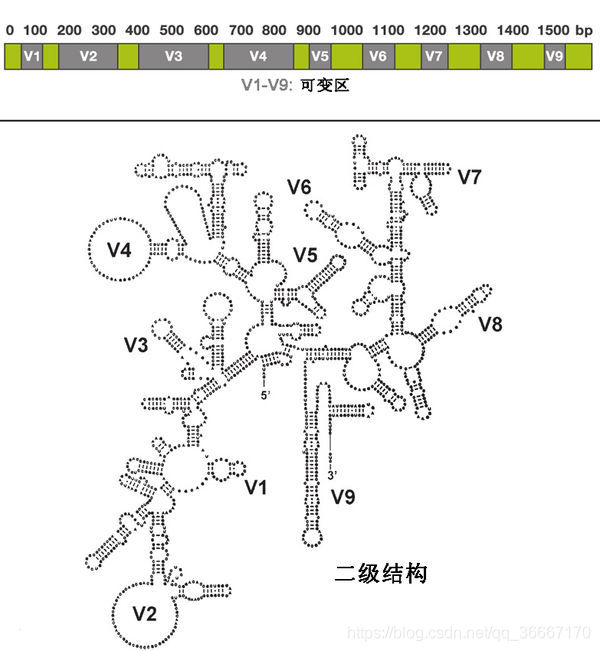
- 分析的长度适宜(约为1540bp):16S rDNA约1.5Kb左右,23S的核苷酸含量过多,几乎是16S核苷酸含量的两倍,分析困难;而5S核苷酸又太少,没有足够的遗传信息,只有16S长度适中信息量较大且易分析。
- 具有良好的进化保守性,16S rDNA的序列包含9个可变区和10个保守区。保守序列区域反映了生物物种间的亲缘关系,而可变序列区域则能体现物种间的差异。
对比16S测序、18S、ITS测序、宏基因组测序
| 16S测序 | 18S/ITS测序 | 宏基因组测序 |
|---|---|---|
| 原核生物的 物种组成、丰度、多样性 | 真核生物的 物种组成、丰度、多样性 | 所有物种(包括病毒) 组成及丰度、基因组成、代谢 |
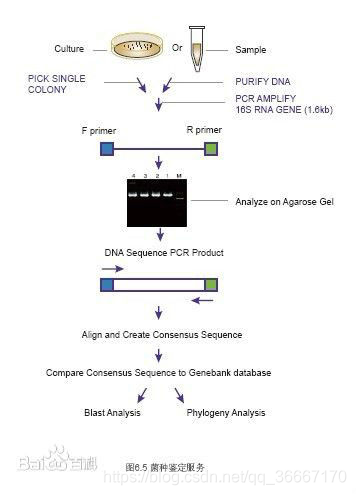
基本步骤
- 细菌基因组提取
- 特异引物扩增16SrDNA序列
- 纯化PCR产物
- DNA测序获得16SrDNA序列
- 与数据库中已知细菌比较获得样品种属信息
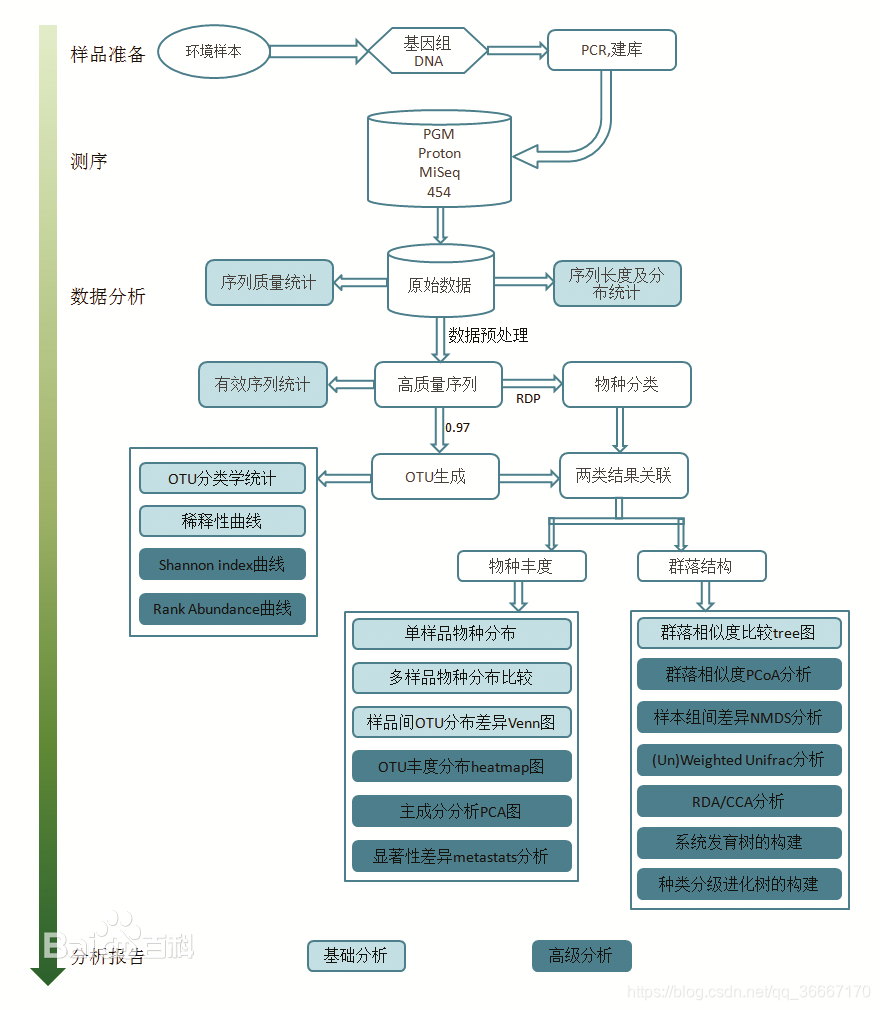
- 数据分析
- 初始数据层面:质量统计,序列长度及分布统计,数据预处理,有效序列统计。
- OTU层面:OTU分类学统计,Alpha多样性分析,稀疏性曲线,Shannon-Wiene曲线,Rank abundance曲线。
- 物种丰度层面:物种分类注释,Beta多样性分析,样本间OTU差异分布分析,OTU丰度分布聚类分析,主成分分析,显著性差异分析,样本组间差异分析。
- 群落结构层面:多样本物种分布比较,群落相似度比较,群落相似度PCoA分析,基于组间进化的差异显著性(Un)Weighted Unifrac分析,RDA/CCA菌群与环境因子之间的关系分析,系统发育树的构建,含种类分级的进化树的构建。
样品提取与测序
样品提取
游离样品
- 土壤,如农田、森林
- 水体,如海洋、污水厂
- 空气,如纽约、新加坡
- 极端,如黄石、冷泉、震区
宿主样品
- 人体,如口腔、肠道、体表
- 动物,如小鼠、大熊猫
- 植物,大豆、小麦
不同的样品提取方法不同,还是要自己查阅相关文献来看怎么提取嗷。
16S测序分析示例问题
- 比较健康与肥胖患者肠道微生物群落结构是否存在差异;
- 清洁空气与雾霾的空气里面包含的微生物是否有差异;
- 土壤覆盖度对微生物群落的影响
16SDNA测序长度的确定
因为16S存在高度保守性和特异性,所以直接就可以使用引物从基因组上扩增出来。转录出来的序列,因为转录水平不同,这个丰度并不能代表物种在群落中的真实丰度。我们最终要分析的是DNA序列,如果用转录的核糖体RNA还得进行反转录,容易出错。
1.设备决定:不同的设备能测量的长度也是不同的,如果设备测序长度为500PE,如下图,V1-V3区域长度为430bp,则一次可以测量V1-V3整块连续区域,同理可以测V3-V5、V4-V6连续区域。图底下给出两种设备的可测读长,Hiseq 2000,150PE可测150PE长度如果用该设备,则只能测V3,以及比V3短的序列;其他设备同理。
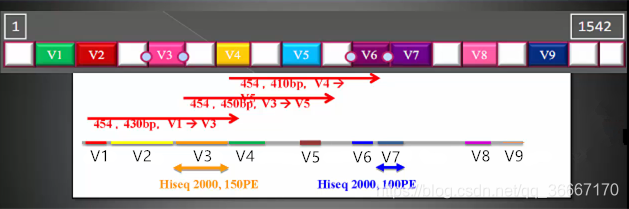
2.常识决定:我们是站在巨人的肩膀上的,经过这么久的积累,通常人们对某些样品要测的大致区域已经有了固定选择:
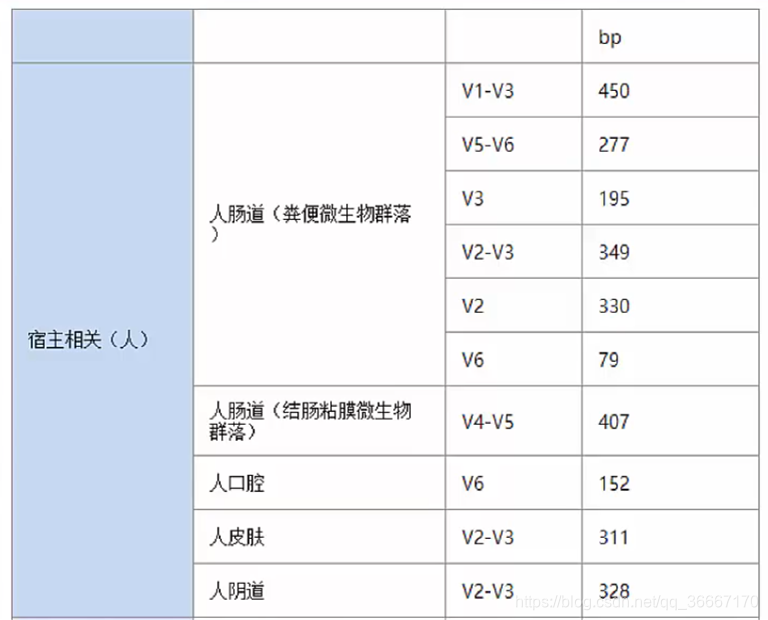
3.根据目前的研究发现,对V3-V4两端进行测序效果最好。当然了,条件允许状况下测序长度越长,准确性越高。
16S测序方法的比较
如何选择测序平台?
1、测序的准确率;
2、通量和价格因素;
3、测序读长;
4、还要考虑数据分析难以程度。
目前常见的设备有Hiseq、Miseq、Pacbio、Life随着科技发展会有更多的平台出现,还是需要权衡之后进行选择。
16S测序分析常用数据库
16S数据库序列部分
测序之后要比对数据库才能知道你测序的样品信息。序列数据库中给每个物种都分配了一个ID,并存储其具有代表性的16S序列,将测序出的结果与数据库进行同源比对,如果比对后结果相似度达到97%才认为达到种间相似性的标准,认为是同种生物。

16S数据库注释信息部分
注释信息部分是ID+分类信息,分类信息具体标注的该ID物种的门、纲、目、科、属、种。当然有的数据库目前还查不到种水平。
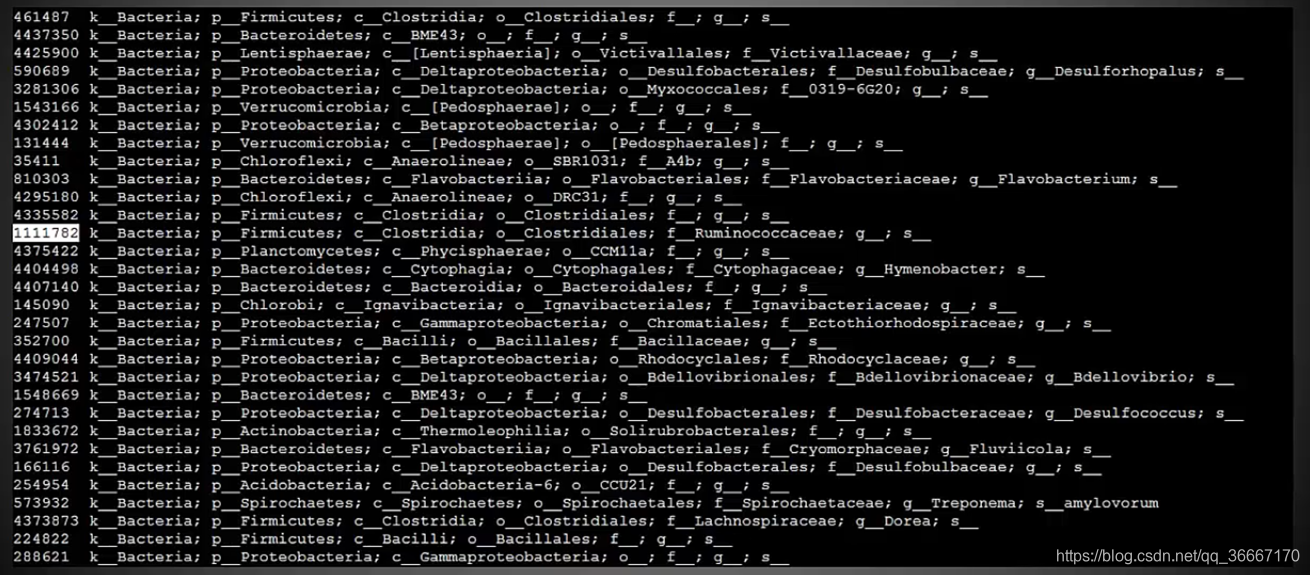
常用数据库推荐
1.https://greengenes.secondgenome.com/

2.https://www.arb-silva.de/

3.http://rdp.cme.msu.edu/

补充
18S
18SrDNA为编码真核生物核糖体小亚基rRNA的DNA序列。对18S rDNA某个高变区进行测序,用于研究环境样本中真核微生物群落结构多样性。
18S分析方法与16S相同,刚才提到的16S数据库也有18S的数据。
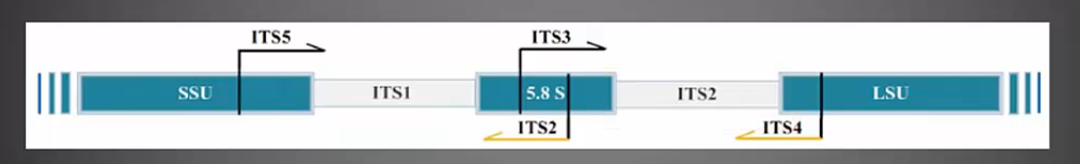
ITS
ITS分为两个区域:ITS1和ITS2,ITS1位于真核生物rDNA序列18S和5.8S之间,ITS2位于真核生物rDNA序列5.8S和28S之间。对ITS1或ITS2进行测序,用于研究环境微生物中真菌群落结构多样性。比18S精确度更高。
常用数据库:
UNITE数据库:User-friendly Nordic ITS Ectomycorrhiza Database
官方网站:http://www2.dpes.qu.se/project/unite/UNITE_intro.htm
HMP计划与EMP计划
这两个自己去了解一下即可。


















 2817
2817

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











