







AWK的名字来自其设计师的首字母:Alfred V. Aho、Peter J. Weinberger和Brian W. Kernighan。
AWK命令可以追溯到Unix早期。它是POSIX标准的一部分,应该可以在任何类unix系统上使用。
虽然与Perl等多用途语言相比,AWK有时会因其过时或缺少特性而受到质疑,但它仍然是我在生信分析中喜欢使用的工具。
本篇教程带你由浅入深,一步步掌握awk命令在生信分析中的重要作用。
声明:
1、 作者水平有限,不足之处请指正!
2、本文不定时持续更新!
AWK commond
AWK程序由一个或多个pattern {action}语句组成。如果对于输入文件的给定记录(“行”),模式计算为非零值(等同于AWK中的“true”),则执行相应操作块中的命令。
1. awk 中常用的预定义变量
RS-记录分隔符
AWK一次只处理一条记录。记录分隔符是用于将输入数据流分割为记录的分隔符。默认情况下,这是换行符。因此,如果不更改它,记录就是输入文件的一行。
NR-当前输入的记录编号
如果正在对记录使用标准的换行分隔符,这将与当前输入行号匹配,即当前行。
FS/OFS-用作字段分隔符的字符
一旦AWK读取了一条记录,它就会根据FS的值将其分割成不同的字段。当AWK在输出中打印一条记录时,它将重新连接字段,但是这次使用OFS分隔符而不是FS分隔符。通常FS和OFS是相同的,但这不是强制性的。“空格”是两者的默认值。
NF-当前记录中的字段数
如果正在为字段使用标准的“空白”分隔符,这将与当前记录中的单词数量匹配。
FILENAME-当前输入文件的名称
当命令行上没有列出数据文件时,awk从标准输入读取,文件名设置为“-”。每次读取新文件时,文件名都会改变。在BEGIN中,FILENAME的值是"",因为还没有处理输入文件。但是,在BEGIN中使用getline可以给FILENAME一个值。
2. awk简单小例子
假设我们有如下bed文件,文件以tab键分隔,包括header,文件一共11行。

- 去除文件header
awk 'NR>1' gene.bed # 等价于
awk 'NR>1{print}' gene.bed # 又等价于
awk 'NR>1{print $0}' gene.bed
由于{print}是AWK使用的默认动作块,$0表示当前行的内容,所以这三条命令得到相同的结果。

- 打印一定区间内的行
# 打印3-6行的基因信息,&&是逻辑运算符“且”
awk 'NR>2&&NR<7' gene.bed

- 删除空行
如果我们数据分析得到的bed文件中包含空行,会给后续的数据处理带来不必要的麻烦,使用awk,去除空行so easy!
加入空格后的bed文件:

awk 'NF' gene.bed

没想到吧,短短的一行命令,使用awk内置的NF变量,轻松删除空行。
- 提取字段
# 提取第一和第二个字段
awk 'BEGIN{FS=OFS="\t"} {print $1,$2}' gene.bed

有空格??加上NF变量去除即可
awk 'BEGIN{FS=OFS="\t"} NF{print $1,$2}' gene.bed

还需要跳过header??一起来试试吧。
awk 'BEGIN{FS=OFS="\t"} NF&&NR>1{print $1,$2}' gene.bed

还不错吧,一步步实现了自身的需求。
- 实现列运算
假设我们有如下txt文件,依然是tab分隔

小需求:统计文件中各类型基因总数
awk 'BEGIN{FS=OFS="\t"} {sum+=$2} END{print sum}' gene_type.txt
# 1842

那么,如果我们有如下文件,我们的需求是统计每一类别基因的数目,如何实现呢?

这里就需要考虑数组了,先展示一下吧,后期会详细讲解。
awk 'BEGIN{FS=OFS="\t"} NR>1{sum[$1]+=$2} END{for(i in sum) print i,sum[i]}' gene_type.txt

3. awk中的数组
数组是AWK的一个强大特性。AWK中的所有数组都是关联数组,因此允许将任意字符串与另一个值关联起来。如果你熟悉其他编程语言,可能会知道数组是散列、关联表、字典或映射。
从表面上看,awk中的数组类似于其他编程语言中的数组,但它们有根本的区别。在awk中,在开始使用数组之前没有必要指定数组的大小。此外,任何数字或字符串,而不仅仅是连续整数,都可以用作数组索引。并且,awk中数组不需要提前声明,这避免了为数组提前分配一个连续的内存块。但是同其他语言一样,awk 索引非负,从0开始。
- 移除重复行
构建文件a,如下:
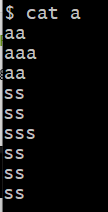
检查文件是否存在重复行,并打印重复行(这里文件较小,肉眼可观察,生信分析中常常要面临上万行,甚至十几万行或者更大的数据)
#打印重复行
awk 'a[$0]++' a
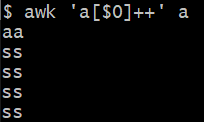
第一次读取记录时,a[$0]是未定义的,因此对于AWK来说等于0。所以第一个记录不会写在输出上。然后这个元素就从0变成了1。简单来说就是,先打印,再递增!
那么,想实现先递增,再打印,如何操作呢?很简单,将a[$0]++改变为++a[$0]即可。
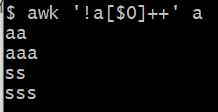
#删除重复行
awk '!a[$0]++' a
来个实例吧,假设我们有如下序列文件,如果我们需要将fasta格式的多行核酸序列转换为一行,应该怎么做?

awk '{if(/^>/) id=$0;else seq[id]=seq[id]$0} END{for(i in seq) print i"\n"seq[i]}' dna.fa

不定时持续更新中!
参考
- 《鸟哥的linux私房菜》
- 维基百科














 870
870











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









