







写在前面:近年来,基于语言模型或逆向折叠算法进行蛋白序列设计可谓如火如荼。然而,预测评估生成的蛋白质是否还会折叠和发挥功能仍具有挑战性。研究人员尝试了20种不同的计算指标,以评估三种模型(祖先序列重建、生成对抗网络和蛋白质语言模型)生成的酶蛋白序列质量。该篇研究重点关注两个酶家族,表达并纯化了500多个天然和生成序列,生成序列与最相似的天然序列具有70-90%一致性。对预测体外酶活性的计算指标进行基准测试。经过三轮实验,该研究提出一种计算过滤标准COMPSS,将实验成功率提高了 50-150%。提出的指标将作为蛋白序列设计的基准,帮助选择活性突变体,从而推动蛋白质工程研究。

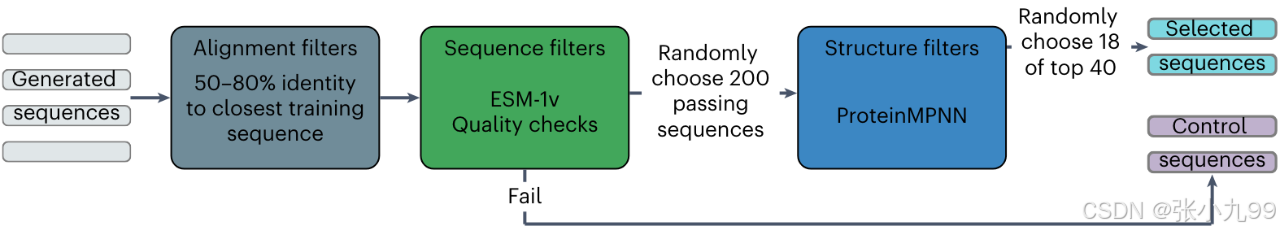
COMPSS框架主要包含以下关键步骤:
-
序列质量检查:确保序列以甲硫氨酸开头,无长重复序列和跨膜结构域。
-
ESM-1v筛选:使用蛋白质语言模型ESM-1v计算序列得分,保留得分高于阈值的序列。
-
AlphaFold2结构预测:对通过ESM-1v筛选的序列进行结构预测。
-
ProteinMPNN打分:使用反向折叠模型ProteinMPNN对预测结构进行打分,选择得分最高的序列进行实验验证。
一、环境的安装
✅ 第一步:克隆代码仓库并进入目录
git clone https://github.com/seanrjohnson/protein_gibbs_sampler.git
cd protein_gibbs_sampler
✅ 第二步:创建 conda 环境并安装依赖
conda env create --name protein_gibbs_sampler -f conda_env.yml✅ 第三步:激活环境
conda activate protein_gibbs_sampler✅ 第四步:测试是否安装成功
pytest . # 该命令会在当前目录运行所有的测试脚本,确保安装无误。注意:第一次执行该命令的时候会自动下载模型权重(esm_msa1b_t12_100M_UR50S.pt),执行时间可能会较长一些
二、部署esm1v蛋白质序列打分模型
1、安装所需的包
原文提供的代码是在需要在colab运行的notebooks脚本,为了使用方便,我们需要将其部署到本地服务器中, 所以需要额外安装一些环境包;
conda install -c conda-forge xlsxwriter
2、测试本地运行,初次执行的时候,需要下载esm系列的模型权重,所以执行速度比较慢,第一次执行成功,下载好模型权重之后,后续执行速度就会变快;
# -*- coding: utf-8 -*-
# @Time : 2025/5/12 20:07
# @Author : Xiaojiu Zhang
# @File : esm1v_score.py
# -*- coding: utf-8 -*-
from glob import glob
import tempfile
import subprocess
import torch
import pandas as pd
import xlsxwriter
import time
import os
# 设置是否启用 ESM-1v 打分
ESM_1v = True
device = 'cuda:0'
results = dict()
start = time.time()
# 输出路径配置
output_excel_path = 'test_esm1v.xlsx'
fasta_files = glob("test.fasta")
# 创建保存结果的函数
def add_metric(metrics_dict, protein_name, metric_name, value):
if protein_name not in metrics_dict:
metrics_dict[protein_name] = dict()
metrics_dict[protein_name][metric_name] = value
# 运行 ESM-1v 无 mask 打分
if ESM_1v:
if device == 'cuda:0':
torch.cuda.empty_cache()
for fasta_file in fasta_files:
with tempfile.TemporaryDirectory() as output_dir:
outfile = os.path.join(output_dir, "esm_results.tsv")
try:
proc = subprocess.run([
'python', "src/pgen/likelihood_esm.py",
"-i", fasta_file,
"-o", outfile,
"--model", "esm1v",
"--masking_off",
"--score_name", "score",
"--device", "gpu"
], stdout=subprocess.PIPE, stderr=subprocess.PIPE, check=True)
print("stdout:", proc.stdout.decode())
print("stderr:", proc.stderr.decode())
except subprocess.CalledProcessError as e:
print("ESM-1v scoring failed:")
print("stdout:", e.stdout.decode())
print("stderr:", e.stderr.decode())
continue # 出错继续下一个文件
# 读取并保存得分结果
df = pd.read_table(outfile)
for _, row in df.iterrows():
add_metric(results, row["id"], "ESM-1v", row["score"])
# 保存结果为 Excel
if results:
os.makedirs(os.path.dirname(output_excel_path), exist_ok=True)
workbook = xlsxwriter.Workbook(output_excel_path)
worksheet = workbook.add_worksheet('sheet1')
worksheet.write(0, 0, "seq")
worksheet.write(0, 1, "ESM-1v")
for i, (seq_id, metrics) in enumerate(results.items(), start=1):
worksheet.write(i, 0, seq_id)
worksheet.write(i, 1, metrics["ESM-1v"])
workbook.close()
print(f"结果已保存至:{output_excel_path}")
else:
print("未生成有效结果,Excel 文件未创建。")
end = time.time()
print(f"计算 {len(results)} 条序列总共耗时:{end - start:.2f} 秒")3、输出结果如下
| seq | ESM-1v |
| UniRef90_A0A017RVR8 | -0.1346586 |
| UniRef90_A0A062U8W6 | -0.1357431 |
| UniRef90_A0A062XZ10 | -0.1139585 |
| UniRef90_A0A077PLI0 | -0.1215153 |
| UniRef90_A0A0A2DZA3 | -0.137109 |
| UniRef90_A0A0C7NZI3 | -0.1371256 |
三、部署ProteinMPNN等结构评估方法
1、安装所需的包
# biotite 是一个用于处理结构生物学数据的 Python 库,支持:解析 .pdb、.mmcif 等结构文件、操作蛋白质结构(坐标、残基、原子等)、序列转换和操作(如 3-letter 到 1-letter)
conda install -c conda-forge biotite
# scipy一个核心科学计算库,它构建在 numpy 的基础之上,提供了大量用于数值计算、科学建模、数学分析和工程问题的高级函数和工具
conda install scipy
# 由于反向折叠模型 GVP 网络需要依赖图神经网络框架 PyTorch Geometric,所以需要安装核心依赖(用 pytorch 2.5 + cu124)
pip install torch-scatter -f https://data.pyg.org/whl/torch-2.5.1+cu124.html
pip install torch-sparse -f https://data.pyg.org/whl/torch-2.5.1+cu124.html
pip install torch-cluster -f https://data.pyg.org/whl/torch-2.5.1+cu124.html
pip install torch-spline-conv -f https://data.pyg.org/whl/torch-2.5.1+cu124.html
# 最后安装主库
pip install torch-geometric2、安装ProteinMPNN
git clone https://github.com/dauparas/ProteinMPNN.git3、测试本地运行
# -*- coding: utf-8 -*-
# @Time : 2025/5/12 15:24
# @Author : Xiaojiu Zhang
# @File : structure_metrics_score.py
# -*- coding: utf-8 -*-
# structure Metrics
# Requires PDB uploaded files. All metrics will execute on all uploaded PDB files.
# The proteinMPNN and ESM-IF scores are the average log likelihood of the query residues. The ProteinMPNN score calculation is performed using the --score_only option of the protein_mpnn_run.py script from the ProteinMPNN repository (https://github.com/dauparas/ProteinMPNN). The ProteinMPNN score is multiplied by -1 so that higher is better, like with other metrics. The ESM-IF score is calculated using the esm.inverse_folding.util.score_sequence function from the ESM repository (https://github.com/facebookresearch/esm). The MIF-ST score is calculated using the extract_mif.py script from the Protein Sequence Models repository (https://github.com/microsoft/protein-sequence-models).
# Average taken from the b-factors in AlphaFold2-produced pdb files.
import os
import torch
import esm
import pandas as pd
from pathlib import Path
from glob import glob
from biotite.structure.io import pdb
from biotite.structure.residues import get_residues
from biotite.sequence import ProteinSequence
import tempfile
import subprocess
import xlsxwriter
import time
# ==== 配置部分 ====
results = dict()
device = 'cuda:0'
pdb_path = "test/*.pdb"
output_pdb_evaluate_excel = "test_structure_metrics.xlsx"
ESM_IF = True # @param {type:"boolean"}
ProteinMPNN = True # @param {type:"boolean"}
MIF_ST = False # @param {type:"boolean"}
AlphaFold2_pLDDT = True # @param {type:"boolean"}
start = time.time()
# ==== 工具函数 ====
def add_metric(metrics_dict, protein_name, metric_name, value):
if protein_name not in metrics_dict:
metrics_dict[protein_name] = dict()
metrics_dict[protein_name][metric_name] = value
def get_pdb_sequence(pdb_path):
with open(pdb_path) as f:
pdb_file = pdb.PDBFile.read(pdb_path)
atoms = pdb_file.get_structure()
residues = get_residues(atoms)[1]
return ''.join([ProteinSequence.convert_letter_3to1(r) for r in residues])
# ==== 计算 ESM-IF 分数 ====
if ESM_IF: # TODO: move to GPU? Maybe spin off into a subprocess when moving to GPU, to avoid memory leaks?
esm_if_model, esm_if_alphabet = esm.pretrained.esm_if1_gvp4_t16_142M_UR50()
esm_if_model.eval()
for pdb_file in glob(pdb_path):
name = Path(pdb_file).stem
coords, seq = esm.inverse_folding.util.load_coords(pdb_file, "A")
ll, _ = esm.inverse_folding.util.score_sequence(esm_if_model, esm_if_alphabet, coords, str(seq))
add_metric(results, name, "ESM-IF", ll)
del esm_if_model, esm_if_alphabet
# ==== 计算 ProteinMPNN 分数 ====
if ProteinMPNN:
print("Running ProteinMPNN scoring...")
with tempfile.TemporaryDirectory() as output_dir:
for i, pdb_file in enumerate(glob(pdb_path)):
name = Path(pdb_file).stem
outfile = output_dir + f"outfile_{i}.txt"
command_line_arguments = [
"python",
"ProteinMPNN/protein_mpnn_run.py",
"--pdb_path", pdb_file,
"--pdb_path_chains", "A",
"--score_only", "1",
"--save_score", "1",
"--out_folder", output_dir,
"--batch_size", "1"
]
with open(outfile, "w") as fh:
proc = subprocess.run(command_line_arguments, stdout=subprocess.PIPE, check=True)
print(proc.stdout.decode('utf-8'), file=fh)
with open(outfile, "r") as score_file_h:
score_file_lines = score_file_h.readlines()
score_line = score_file_lines[-2].split(",")
score_parts = score_line[1].strip().split(": ")
assert score_parts[0] == "mean"
score = -1 * float(score_parts[1])
add_metric(results, name, "ProteinMPNN", score)
# ==== 计算 AlphaFold2 pLDDT 分数 ====
if AlphaFold2_pLDDT:
for pdb_file in glob(pdb_path):
name = Path(pdb_file).stem
pdb_file_obj = pdb.PDBFile.read(pdb_file)
atoms = pdb_file_obj.get_structure(extra_fields=['b_factor'])
prev_residue = -1
plddt_sum = 0
residue_count = 0
for a in atoms[0]:
if a.res_id != prev_residue:
prev_residue = a.res_id
residue_count += 1
plddt_sum += a.b_factor
plddt_avg = plddt_sum / residue_count if residue_count else 0
add_metric(results, name, "AlphaFold2_pLDDT", plddt_avg)
# ==== 保存 CSV 和 Excel 结果 ====
df = pd.DataFrame.from_dict(results, orient="index")
df.to_csv("calculated_metrics.csv")
workbook = xlsxwriter.Workbook(output_pdb_evaluate_excel)
worksheet = workbook.add_worksheet('sheet1')
headers = ["seq", "ESM-IF", "ProteinMPNN", "AlphaFold2_pLDDT"]
for col, header in enumerate(headers):
worksheet.write(0, col, header)
print(df)
for row, (key, value) in enumerate(results.items(), start=1):
worksheet.write(row, 0, key)
worksheet.write(row, 1, value.get("ESM-IF", ""))
worksheet.write(row, 2, value.get("ProteinMPNN", ""))
worksheet.write(row, 3, value.get("AlphaFold2_pLDDT", ""))
workbook.close()
end = time.time()
print(f"\n✅ 共处理 {len(results)} 个结构,耗时 {end - start:.2f} 秒")4、输出结果如下
| seq | ESM-IF | ProteinMPNN | AlphaFold2_pLDDT |
| UniRef90_A0A017RVR8 | -1.34865 | -1.6513 | 73.60916 |
| UniRef90_A0A062U8W6 | -1.43547 | -1.7539 | 73.18308 |
| UniRef90_A0A062XZ10 | -1.12485 | -1.4414 | 83.58752 |
| UniRef90_A0A077PLI0 | -1.20124 | -1.5233 | 78.54388 |
| UniRef90_A0A0A2DZA3 | -1.03581 | -1.465 | 85.55203 |
| UniRef90_A0A0C7NZI3 | -1.22971 | -1.5248 | 85.40533 |
总结:COMPSS 的核心思想是通过生物学驱动的序列质量检查和蛋白质语言模型评分进行预过滤来选择序列,最后利用相对耗时的结构预测进行最终评分。
参考文档:
GitHub - seanrjohnson/protein_gibbs_sampler: Gibbs sampling for generating protein sequences


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









