







一、在线使用
如果使用次数不多或者计算资源不足的用户来说,ProteinMPNN也提供了Huggingface和colab在线使用的方法,链接如下:
1、ProteinMPNN - a Hugging Face Space by simonduerr
https://huggingface.co/spaces/simonduerr/ProteinMPNN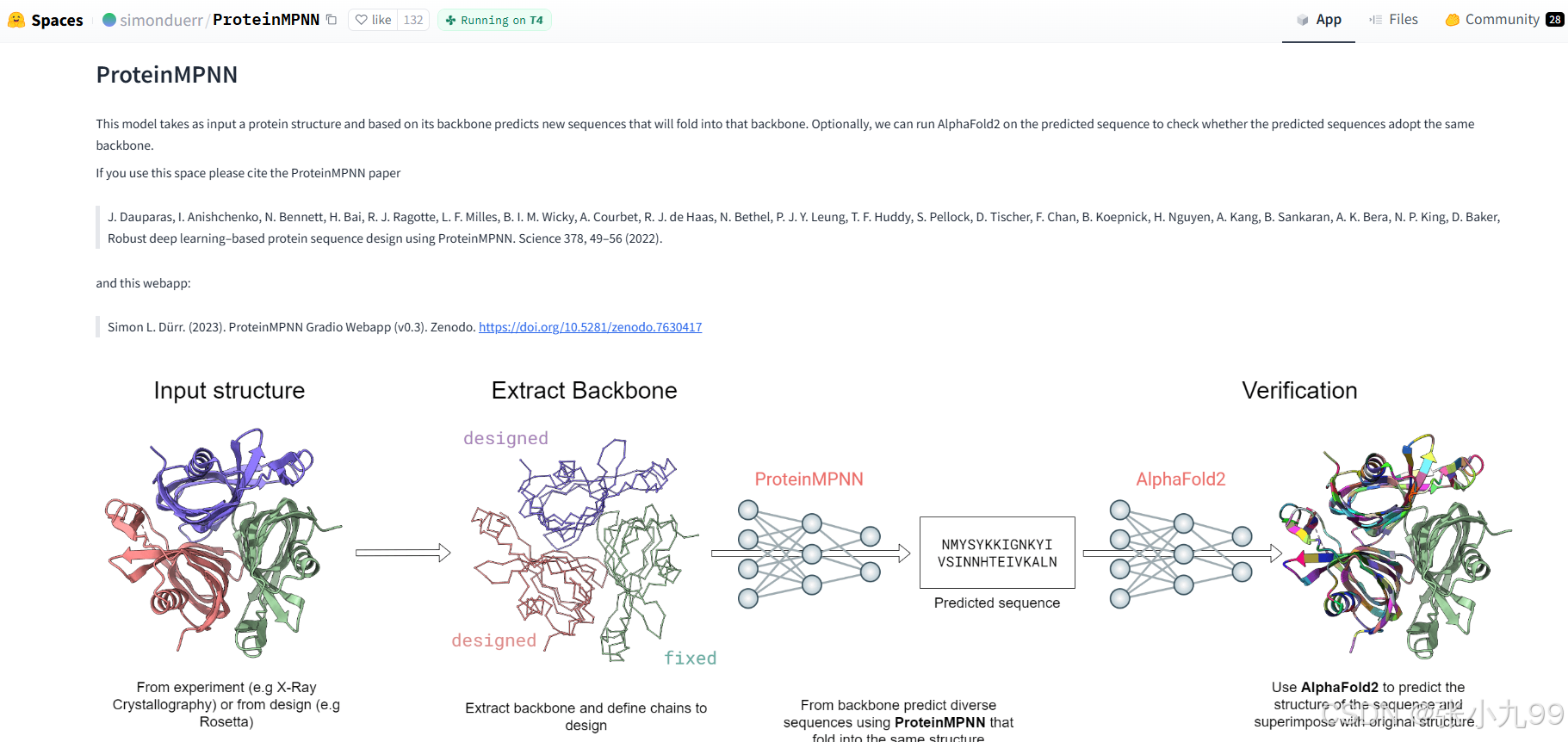
2、Colaboratory
https://colab.research.google.com/github/dauparas/ProteinMPNN/blob/main/colab_notebooks/quickdemo.ipynb
二、 本地安装
1、首先从github将源代码下载到服务器
git clone https://github.com/dauparas/ProteinMPNN.git # 克隆仓库到本地
cd ProteinMPNN # 进入ProteinMPNN的项目文件2、创建虚拟环境
conda create --name proteinMPNN # 创建名字为ProteinMPNN的虚拟环境
conda activate proteinMPNN # 激活该环境
conda install pytorch torchvision torchaudio cudatoolkit=11.3 -c pytorch # 安装PyTorch、TorchVision、Torchaudio 以及 CUDA 11.3 工具包(确保服务器中有NVIDIA GPU,否则可能会自动安装CPU版本)注意:本来执行上述指令安装最新的pytorch,但是发现上面的指令执行过后安装的是cpu支持的torch,所以下面这个指令制定了pytorch的具体版本,这样安装的就是GPU支持的了。
原因:如果没有明确指定 PyTorch 的版本,Conda 会选择最新的可用版本。PyTorch 的最新版本(比如 PyTorch 2.5.1)。但是默认安装最新版本的 PyTorch 时,可能会出现与指定的 CUDA工具包(cudatoolkit=11.3) 不兼容的情况,从而导致 安装了 CPU 版本的 PyTorch。可以去pytorch官网查找支持所需cuda版本的pytorch版本(Previous PyTorch Versions | PyTorch)
conda install pytorch==1.12.1 torchvision==0.13.1 torchaudio==0.12.1 cudatoolkit=11.3 -c pytorch
3、 运行测试脚本
(1)运行以下命令显示主ProteinMPNN应用程序的帮助信息:
cd ProteinMPNN #cd 到下载好的ProteinMPNN的文件夹
python protein_mpnn_run.py -h # 该命令将显示脚本使用说明和可用的输入参数。该脚本是初始化和运行模型的唯一脚本。其他可用的函数用于准备输入数据和创建选项文件。输入参数如下:
argparser.add_argument("--suppress_print", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--ca_only", action="store_true", default=False, help="Parse CA-only structures and use CA-only models (default: false)")
argparser.add_argument("--path_to_model_weights", type=str, default="", help="Path to model weights folder;")
argparser.add_argument("--model_name", type=str, default="v_48_020", help="ProteinMPNN model name: v_48_002, v_48_010, v_48_020, v_48_030; v_48_010=version with 48 edges 0.10A noise")
argparser.add_argument("--use_soluble_model", action="store_true", default=False, help="Flag to load ProteinMPNN weights trained on soluble proteins only.")
argparser.add_argument("--seed", type=int, default=0, help="If set to 0 then a random seed will be picked;")
argparser.add_argument("--save_score", type=int, default=0, help="0 for False, 1 for True; save score=-log_prob to npy files")
argparser.add_argument("--path_to_fasta", type=str, default="", help="score provided input sequence in a fasta format; e.g. GGGGGG/PPPPS/WWW for chains A, B, C sorted alphabetically and separated by /")
argparser.add_argument("--save_probs", type=int, default=0, help="0 for False, 1 for True; save MPNN predicted probabilites per position")
argparser.add_argument("--score_only", type=int, default=0, help="0 for False, 1 for True; score input backbone-sequence pairs")
argparser.add_argument("--conditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output conditional probabilities p(s_i given the rest of the sequence and backbone)")
argparser.add_argument("--conditional_probs_only_backbone", type=int, default=0, help="0 for False, 1 for True; if true output conditional probabilities p(s_i given backbone)")
argparser.add_argument("--unconditional_probs_only", type=int, default=0, help="0 for False, 1 for True; output unconditional probabilities p(s_i given backbone) in one forward pass")
argparser.add_argument("--backbone_noise", type=float, default=0.00, help="Standard deviation of Gaussian noise to add to backbone atoms")
argparser.add_argument("--num_seq_per_target", type=int, default=1, help="Number of sequences to generate per target")
argparser.add_argument("--batch_size", type=int, default=1, help="Batch size; can set higher for titan, quadro GPUs, reduce this if running out of GPU memory")
argparser.add_argument("--max_length", type=int, default=200000, help="Max sequence length")
argparser.add_argument("--sampling_temp", type=str, default="0.1", help="A string of temperatures, 0.2 0.25 0.5. Sampling temperature for amino acids. Suggested values 0.1, 0.15, 0.2, 0.25, 0.3. Higher values will lead to more diversity.")
argparser.add_argument("--out_folder", type=str, help="Path to a folder to output sequences, e.g. /home/out/")
argparser.add_argument("--pdb_path", type=str, default='', help="Path to a single PDB to be designed")
argparser.add_argument("--pdb_path_chains", type=str, default='', help="Define which chains need to be designed for a single PDB ")
argparser.add_argument("--jsonl_path", type=str, help="Path to a folder with parsed pdb into jsonl")
argparser.add_argument("--chain_id_jsonl",type=str, default='', help="Path to a dictionary specifying which chains need to be designed and which ones are fixed, if not specied all chains will be designed.")
argparser.add_argument("--fixed_positions_jsonl", type=str, default='', help="Path to a dictionary with fixed positions")
argparser.add_argument("--omit_AAs", type=list, default='X', help="Specify which amino acids should be omitted in the generated sequence, e.g. 'AC' would omit alanine and cystine.")
argparser.add_argument("--bias_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies AA composion bias if neededi, e.g. {A: -1.1, F: 0.7} would make A less likely and F more likely.")
argparser.add_argument("--bias_by_res_jsonl", default='', help="Path to dictionary with per position bias.")
argparser.add_argument("--omit_AA_jsonl", type=str, default='', help="Path to a dictionary which specifies which amino acids need to be omited from design at specific chain indices")
argparser.add_argument("--pssm_jsonl", type=str, default='', help="Path to a dictionary with pssm")
argparser.add_argument("--pssm_multi", type=float, default=0.0, help="A value between [0.0, 1.0], 0.0 means do not use pssm, 1.0 ignore MPNN predictions")
argparser.add_argument("--pssm_threshold", type=float, default=0.0, help="A value between -inf + inf to restric per position AAs")
argparser.add_argument("--pssm_log_odds_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--pssm_bias_flag", type=int, default=0, help="0 for False, 1 for True")
argparser.add_argument("--tied_positions_jsonl", type=str, default='', help="Path to a dictionary with tied positions")
(2)测试脚本:在没有附加选项的情况下对1QYS的整个序列进行简单的重新设计。切换到教程目录并创建一个新目录以重新设计1QYS序列。
mkdir 0_output_test/1_basic_usage/input # 创建输入目录
mkdir 0_output_test/1_basic_usage/output # 创建输出目录
python protein_mpnn_run.py --pdb_path 0_output_test/1_basic_usage/input/5L33.pdb --out_folder 0_output_test/1_basic_usage/output/5L33 --num_seq_per_target 10 --sampling_temp "0.1" --seed 9 --batch_size 1 --model_name v_48_020 # 无条件对5L33的序列进行重设计
--pdb_path输入pdb文件的全路径;
--pdb_path_chains需要设计pdb文件中的哪一条链;
--out_folder文件输出路径;
--num_seq_per_target设计的蛋白序列输出条数;
--sampling_temp采样温度,建议可取值0.1, 0.15, 0.2, 0.25, 0.3;
--seed设置随机数
--batch_size批大小
--model_name设置要使用的 ProteinMPNN 的模型名称
如果报下述错误,原因是当前NumPy 2.2.3与依赖的模块不兼容,需降级到NumPy 1.x
# 若使用conda环境,先激活环境
conda activate proteinMPNN
# 降级NumPy到1.23.5(推荐稳定版本)
pip uninstall numpy -y
pip install numpy==1.23.5
# 验证安装版本
python -c "import numpy; print(numpy.__version__)"
# 应输出:1.23.5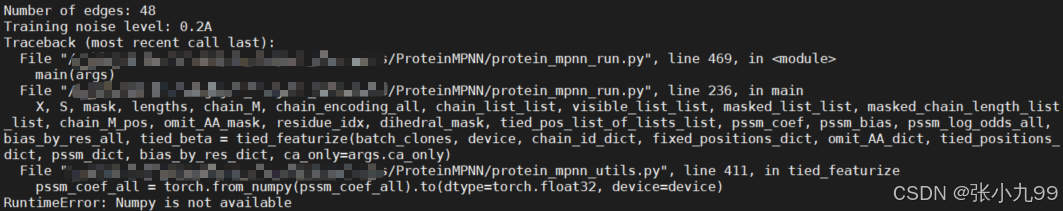
查看生成的序列文件:
gedit ./0_output_test/output/5L33/seqs/5L33.fa5L33.fa包含原始序列以及fasta格式中的所有设计。每个序列都附带一个分数和一个序列恢复值。在这种情况下,分数和全局分数应该是相同的。全局分数对序列中的所有残基进行平均,而分数仅代表设计残基的平均值。序列恢复显示设计残基的百分比identity。
三、 任务示例
1、无条件生成
python protein_mpnn_run.py --pdb_path 0_test_demo/1_basic_usage/input/1qys.pdb --out_folder 0_test_demo/1_basic_usage/output/1qys --num_seq_per_target 10 --sampling_temp "0.1" --seed 9 --batch_size 1 --model_name v_48_020 # 从头设计1qys的序列1qys.fa包含原始序列以及fasta格式中的所有设计序列,如下所示:
>1qys, score=1.7137, global_score=1.7137, fixed_chains=[], designed_chains=['A'], model_name=v_48_020, git_hash=8907e6671bfbfc92303b5f79c4b5e6ce47cdef57, seed=9
DIQVQVNIDDNGKNFDYTYTVTTESELQKVLNELMDYIKKQGAKRVRISITARTKKEAEKFAAILIKVFAELGYNDINVTFDGDTVTVEGQL
>T=0.1, sample=1, score=0.8666, global_score=0.8666, seq_recovery=0.4457
GIKITVTIEDGDETEIIEYEVESEEELEKVLEEIKKIIKEKNPKKVTISVEAETPEEAKKYAEKLKKLLKELGYNDIEVKMEGNTVTVTGTK
>T=0.1, sample=2, score=0.8723, global_score=0.8723, seq_recovery=0.3913
MIKIKVVIEKGDEKIELEYEVESEEELEKVLEEIKKLIKEKNPEKVTISVEAETPEKAKEYAEKLEKLFKELGYSEIEVKFEGNKVTVTGKR
>T=0.1, sample=3, score=0.8718, global_score=0.8718, seq_recovery=0.4348
MIKITVTIEDGDKTEVIEETVESDEELKEVLEKLKEKIKKANPEKVTISVTAETPEKAKEYAEKLKKLLTELGYKDITVEFDGNTVTVTGKK
>T=0.1, sample=4, score=0.9126, global_score=0.9126, seq_recovery=0.4239
MIKIKVVIEKGDKKIEIEKEVESKEEFEKVLKELKELIKKLDPEKVTISVTAETPEEAEEYAKKLREFFEELGYKDIEVKLEGNTVTVTGKK
>T=0.1, sample=5, score=0.8440, global_score=0.8440, seq_recovery=0.4239
MIEITVIIEKDNKKIEIKEEVESEEELEEVLERIKKIIKEENPEKVTISIEAETKEEAEKYAEKLKKLLEELGYKDIKVEFEGNKVTVTGKK
>T=0.1, sample=6, score=0.8459, global_score=0.8459, seq_recovery=0.4783
MIKITVTIEKGDKTIKLEYEVDSEEEFKKVLEEIKKIIKEENPEKVTISVEAETKEEAEKYAKILKKLFEELGYKDIKVEFDGNTVTVTGTK
>T=0.1, sample=7, score=0.8921, global_score=0.8921, seq_recovery=0.4674
MIKITVTIEDGDETIVLEYEVESEEEFKKVLEEIKEIIKEKNPKVVTISVKAETKEEAEKYAKILKELLTELGYNDIKVEFNGNTVTVTGTK
>T=0.1, sample=8, score=0.8632, global_score=0.8632, seq_recovery=0.4348
MIKIKVTIEKGNEKEVLEYEVESKEELKEVLEEIEKIIKEKNPEEVTISVTAKTKEEAEEYAEILKKLLKKLGYKDIEVKFDGNTVTVTGKK
>T=0.1, sample=9, score=0.8514, global_score=0.8514, seq_recovery=0.4783
MIKITVKIEKDNKTIELEYEVESEEEFEKVLEEIKEIIKKENPEKVTISVEAETKEEAEKYAEKLKKLFEELGYNDIKVEFNGNTVTVTGKK
>T=0.1, sample=10, score=0.8598, global_score=0.8598, seq_recovery=0.4348
MIKIKVTIEKGDEKEVLEYEVESEEELEEVLKEIEKIIKEKNPEKVTISVTAKTPEEAKKYAERLKKLFKKLGYNNITVEFDGNTVTVTGEK
2、残基偏好
在ProteinMPNN/helper_scripts/中可以找到用于生成输入文件以使用ProteinMPNN的一些专用选项的脚本(残基偏差,省略氨基酸,固定位置)。其中一个应用是引入残基偏向,例如使疏水残基更有可能而半胱氨酸更不可能。
mkdir 0_test_demo/2_residue_bias # 创建路径
mkdir 0_test_demo/2_residue_bias/input # 创建输入文件路径
mkdir 0_test_demo/2_residue_bias/output # 创建输出文件路径
python helper_scripts/make_bias_AA.py --output_path 0_test_demo/2_residue_bias/input/1qys_bias.jsonl --AA_list "A F G I L M P V W Y C" --bias_list "1.39 1.39 1.39 1.39 1.39 1.39 1.39 1.39 1.39 1.39 -1.1" # 设置残基偏好权重
python protein_mpnn_run.py --pdb_path 0_test_demo/2_residue_bias/input/1qys.pdb --out_folder 0_test_demo/2_residue_bias/output --num_seq_per_target 10 --sampling_temp "0.1" --seed 9 --batch_size 1 --model_name v_48_020 --bias_AA_jsonl 0_test_demo/2_residue_bias/1qys_bias.jsonl # 使用残基偏好重新设计1qys序列
生成的序列如下所示:
>1qys, score=1.7137, global_score=1.7137, fixed_chains=[], designed_chains=['A'], model_name=v_48_020, git_hash=8907e6671bfbfc92303b5f79c4b5e6ce47cdef57, seed=9
DIQVQVNIDDNGKNFDYTYTVTTESELQKVLNELMDYIKKQGAKRVRISITARTKKEAEKFAAILIKVFAELGYNDINVTFDGDTVTVEGQL
>T=0.1, sample=1, score=1.1176, global_score=1.1176, seq_recovery=0.3478
GVLVVVVIVDGGAVEVLVDVVAGDAALAAVLAALAARIAAAAPPVVTIAVTAATPAAAAAYAARLRALLAALGYADIEVALEGNTVIVTGRR
>T=0.1, sample=2, score=1.1237, global_score=1.1237, seq_recovery=0.3478
GIAVTVVLEAGGLRLVLRAAVAGDAALAAVLAALAALIAALAPPVVTVAVAAATPAAALALAARLRALFAALGYADLAVALDGTTVTVTGRL
>T=0.1, sample=3, score=1.1754, global_score=1.1754, seq_recovery=0.3913
AVVVTVVIEDGGAVAVYNYVVAGAAALAAVLAALAAVIAAANPATVTISVTAATPAAAAIYAAALTALLTALGYTDITVEMDGTTVTVTGVK
>T=0.1, sample=4, score=1.2606, global_score=1.2606, seq_recovery=0.3587
AVLVTVVLVAGGAVLVLRAVVAGDAALAAVLAALAALIAALAPPVVAVAVTAATPAAAAVYAARLLALFAALGYADIAVALDGTTVTVTGRL
>T=0.1, sample=5, score=1.2502, global_score=1.2502, seq_recovery=0.3587
ALVVRVVIEKGGETIVLEWTVAGAAALAAVLAAVAAIIAALAPPVVTIAVTAATPAAAAVYAAQLLALLAALGYADITVAFAGTTVVVTGRL
>T=0.1, sample=6, score=1.2929, global_score=1.2929, seq_recovery=0.4022
AVVVLVVIEDGGLVIELRYVVAGDAAFAAVLAAIAALIAALAPPRVTIAVTAATPAAAIAYAAILRALFAALGYADIEVVLDGTTVTVTGVL
>T=0.1, sample=7, score=1.1509, global_score=1.1509, seq_recovery=0.3370
AVAVTVVIEAGGAVVVVEAVVAGAAALAAVLAALAALIAAAAPPVVTIAVTAATAAAAAAYAAVLTALLAALGYADLTVALAGTTVTVTGVR
>T=0.1, sample=8, score=1.1386, global_score=1.1386, seq_recovery=0.3478
ALVVTVVIAAGAATAVAVAVVAGDAALAAVLAALAAAIAAAAPPVVTIAVTAATPAAAAAYAAALTALLAALGYADIAVAFAGTTVTVTGRR
>T=0.1, sample=9, score=1.2726, global_score=1.2726, seq_recovery=0.3913
GVLILVVIEKGGLVIVLLYSVAGAAALAAVLAAIAALIKALAPPRVTIAVTAATPAAAAAYAAQLLALFKALGYKDIEVVFDGNTVTVTGVL
>T=0.1, sample=10, score=1.1829, global_score=1.1829, seq_recovery=0.3696
AITITVVIEAGGATVVLVAVVAGAAALAAVLAALAALIAAAAPPRVTIAVTAATPAAAAAYAALLRALFAALGYADLTVALDGTTVTVTGVR
3、固定位置
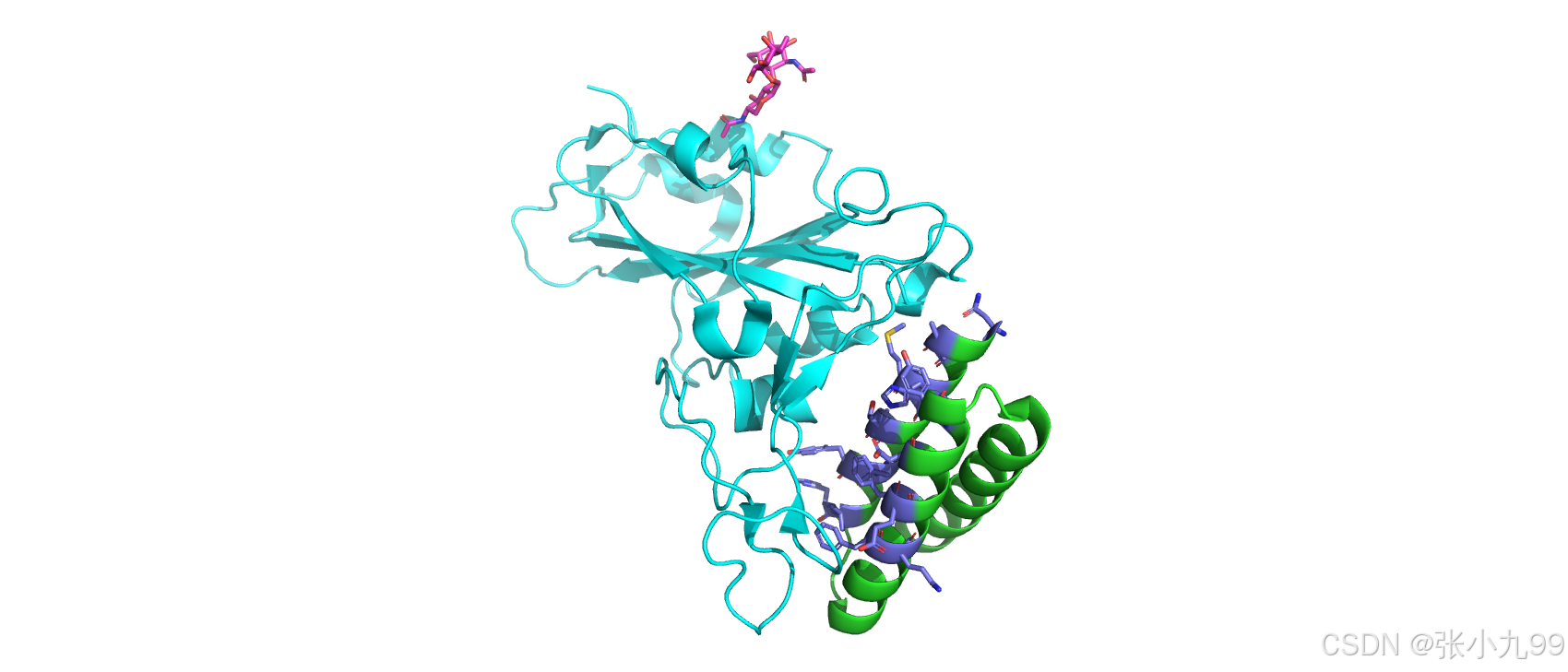
(1)从实际的科学研究角度来看,仅重新设计蛋白质中的某些位置并保持其他部分固定可能是有意义的。在下一个案例中,我们想要重新设计SARS-CoV-2刺突蛋白受体结合结构域和迷你蛋白抑制剂LCB3之间的界面。该复合物的结构在7JZM下被结晶并发表。我们只想设计LCB3(链A)的残基。为此,我们首先为包含我们要重新设计位置的链分配链。
# 提取PDB文件的氨基酸序列和坐标信息,提取所有链中所有残基的 N, CA, C, O原子的坐标,并且把他们的信息存储为字典,保存到JSON格式的输出文件中
python helper_scripts/parse_multiple_chains.py --input_path 0_test_demo/3_fixing_positions/input --output_path 0_test_demo/3_fixing_positions/input/7jzm.jsonl
# 从输入的 JSON 文件中读取多个蛋白质序列数据,并根据用户提供的链列表,标识哪些链需要设计(designed_chain_list),哪些链不需要设计(fixed_chain_list)。然后,它将这些信息以字典的形式保存到指定的输出文件中。
# 示例输出:{"7jzm": [["A"], ["B"]]},A 是需要设计的链,B 是不需要设计的链。
python helper_scripts/assign_fixed_chains.py --input_path 0_test_demo/3_fixing_positions/input/7jzm.jsonl --output_path 0_test_demo/3_fixing_positions/input/7jzm_assigned.jsonl --chain_list "A" (2)假设LCB3的界面残基包括与刺突蛋白的距离为5埃的所有残基。使用pymol检查蛋白质结构并选择要进行设计的残基是一种方式。在这种情况下,我们在pymol中加载结构:
pymol 0_test_demo/3_fixing_positions/input/7jzm.pdb(3)选择并输出LCB3界面的残基。我们在pymol命令行中输入以下内容:
select interface, br. chain A within 5 of chain B # 该命令选择链 A 中与链 B 相互作用的部分,距离链 B 中任意原子小于或等于 5 Å 的区域,并命名为 interface。br. 是 byresidue(按残基选择)的缩写,它表示选择时会基于 残基(而不是原子)进行。
reslist =[]
iterate interface and name CA, reslist.append((resi)) # 迭代 interface 区域中的每个残基,只考虑 Cα 原子,并将每个残基的编号添加到 reslist 列表中
print(*reslist) # 打印 reslist 列表中的所有残基编号(4)如下图所示,选择需要设计的残基是两个蛋白的界面区域,即图中的紫色区域:
(5)假如现在有一个位置列表,可以将其设置为可设计。ProteinMPNN只能接受应该固定的位置字典。使用以下python脚本创建包含固定位置的jsonl文件,以字典格式。
# --specify_non_fixed的值为True,表明在链 A 中,position_list 中的残基是需要设计的,而其他链的其他残基会被视为固定的。
python helper_scripts/make_fixed_positions_dict.py --specify_non_fixed --position_list "1 3 4 6 7 8 10 11 13 14 17 18 30 31 33 34 37 40" --chain_list "A" --input_path 0_test_demo/3_fixing_positions/input/7jzm.jsonl --output_path 0_test_demo/3_fixing_positions/input/7jzm_fixed_pos.jsonl 上述 make_fixed_positions_dict.py 函数的一些参数说明:
input_path:输入JSON文件路径,每行一个JSON对象。
output_path:输出配置文件路径。
chain_list:需要操作的链列表(如"A B")。
position_list:各链的固定/设计位置列表(如"11 12, 1 2")。
specify_non_fixed:标志位,若为True,则position_list指定的是需要设计的残基,而非固定。默认为False,即position_list指定的是需要固定的残基。最终的输出的 JSON 文件(7jzm_fixed_pos.jsonl)会包含每个链的固定位置,不在文件中的点被认为是模型设计的位置。
(6)使用特定的残基运行ProteinMPNN,这次设计只会对选择的位置进行设计,其他位置固定不变。此外,这次我们想输出每个残基的得分,以了解哪些点突变对序列得分贡献最大。
python protein_mpnn_run.py --jsonl_path 0_test_demo/3_fixing_positions/input/7jzm.jsonl --chain_id_jsonl 0_test_demo/3_fixing_positions/input/7jzm_assigned.jsonl --fixed_positions_jsonl 0_test_demo/3_fixing_positions/input/7jzm_fixed_pos.jsonl --out_folder 0_test_demo/3_fixing_positions/output --num_seq_per_target 10 --sampling_temp "0.1" --seed 9 --batch_size 1 --save_score 1 --save_probs 1
save_score:控制是否保存-log(probability)作为得分。
--save_probs:控制是否保存 MPNN 模型预测的每个位置的概率。
(7)生成的序列如下所示:
>7jzm, score=1.8346, global_score=1.5371, fixed_chains=['B'], designed_chains=['A'], model_name=v_48_020, git_hash=8907e6671bfbfc92303b5f79c4b5e6ce47cdef57, seed=9
NDDELHMLMTDLVYEALHFAKDEEIKKRVFQLFELADKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=1, score=0.9824, global_score=1.4605, seq_recovery=0.4444
NDNELWMEMNDLVYEALKFAKDEEIKKRVYELYELALKAHKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=2, score=0.9898, global_score=1.4564, seq_recovery=0.4444
NDNELWQEMNDLVYEALKFAKDEEIKKRVNELYELALKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=3, score=1.0127, global_score=1.4588, seq_recovery=0.4444
NDNELWQEMLDLVYEALKFAKDEEIKKRVNELYELAEKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=4, score=0.9453, global_score=1.4493, seq_recovery=0.4444
NDNELWQEMNDLVYEALKFAKDEEIKKRVYELYELARKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=5, score=0.9510, global_score=1.4566, seq_recovery=0.4444
NDNELWQEMNDLVYEALKFAKDEEIKKRVYELYELARKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=6, score=0.9538, global_score=1.4389, seq_recovery=0.4444
NDNELWAEMNDLVYEALKFAKDEEIKKRVYELYELARKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=7, score=0.9373, global_score=1.4433, seq_recovery=0.4444
NDNELWQEMNDLVYEALKFAKDEEIKKRVYELYELALKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=8, score=0.9219, global_score=1.4770, seq_recovery=0.4444
NDNELWQEMNDLVYEALKFAKDEEIKKRVYELYELARKAYKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=9, score=1.0416, global_score=1.4601, seq_recovery=0.3889
NDNELWQEMNDLVYEALKFAKDEEIKKRVNELYELALKARKNNDRQKLEKVVEELKELLERLLS
>T=0.1, sample=10, score=0.9328, global_score=1.4467, seq_recovery=0.4444
NDNELWQEMLDLVYEALKFAKDEEIKKRVYELYELARKAYKNNDRQKLEKVVEELKELLERLLS
一些参数的说明如下:
- score - 在设计的残基上取平均值,即采样的氨基酸的负对数概率。
- global score - 在所有链上所有残基的平均值,即采样的氨基酸和固定氨基酸的负对数概率。
- fixed_chains - 没有被设计的链(固定链)。
- designed_chains - 被重新设计的链。
- model_name/CA_model_name - 用于生成结果的模型名称,例如
v_48_020。- git_hash - 用于生成输出的 GitHub 版本。
- seed - 随机种子。
- T=0.1 - 使用温度为 0.1 的采样方法生成序列。
- sample - 序列的采样编号 1, 2, 3...等。
- seq_recovery - 即序列恢复率,指的是模型生成的蛋白序列中,与原始参考序列相比,氨基酸完全匹配的位置所占的比例。
注意:
1、这两种得分,score 和 global score,都是通过计算氨基酸的 负对数概率(即
-log(probability))来衡量模型生成的序列的质量。负对数概率越小,意味着序列的概率越高,因此,得分越低表示模型生成的序列越有可能是自然的或合理的。2、现在,序列分数和全局分数应该有所不同,因为这次我们只设计了蛋白质的一部分。
(8)结果分析及其可视化(可选):
安装JupyterLab库,JupyterLab是一种用于编码的交互式界面,允许我们轻松更改和运行代码。
# 安装 Jupyter
conda install jupyterlab在Jupyter notebook会话中,我们可以交互地绘制氨基酸概率。可以通过运行以下命令启动notebook。
jupyter lab scripts/plot_probs.ipynb # 打开plot_probs文件通过单击相应字段中的播放/运行按钮或按Ctrl+Enter键执行两个脚本块。第一个块绘制了复合物所有残基的输出氨基酸概率。在第二个块中,我们指定了重新设计的位置。现在,我们可以调查我们感兴趣的每个位置的单个分数。
注意:目前最新下载的github源码文件中并没有上述脚本,根据经验,可以在ProteinMPNN/colab_notebooks/quickdemo.ipynb 路径下的ipynb文件中运行并绘制结果。


















 1292
1292

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









