






 本文深入探讨了短串联重复序列(STR)基因分型技术,介绍了STR的基本概念、遗传特性及其在个体识别中的应用。文章详细解释了通过PCR和NGS方法进行STR检测的过程,并提供了HipSTR等软件的使用指南,最后指出了STR数据库资源。
本文深入探讨了短串联重复序列(STR)基因分型技术,介绍了STR的基本概念、遗传特性及其在个体识别中的应用。文章详细解释了通过PCR和NGS方法进行STR检测的过程,并提供了HipSTR等软件的使用指南,最后指出了STR数据库资源。
人类基因组DNA有30亿个碱基(bp),其中10%是串联重复序列,称为卫星DNA。按重复单位的长短,又可分为大卫星、中卫星、小卫星和微卫星。
STR: 短串联重复序列(short tandem repeats,STR)也称微卫星DNA(microsatellite DNA), 通常是基因组中由1~6个碱基单元组成的一段DNA重复序列。STR序列符合孟德尔遗传定律,个体间存在相同的短串联重复序列,但重复的次数在个体间存在差异,形成片段长度不等的等位基因。由于核心单位重复数目在个体间呈高度变异性并且数量丰富,构成了STR基因座的遗传多态性。
通常STR检测是对基因组特定STR区域进行PCR扩增,电泳,然后进行STR分型鉴定。如下图所示,当一个STR出现单峰时,说明该个体在该STR区域,为纯合,出现双峰则说明两条染色体的STR重复次数不同,为杂合。
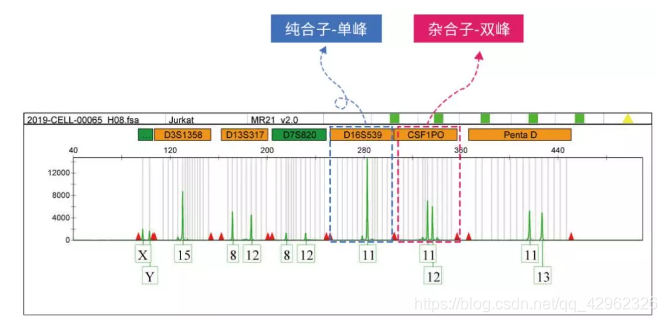
一般情况下,每一个STR位点会被约5-20%的人们所共有,当同时鉴别多个STR位点时,通过最终的STR图谱可以非常精确地鉴别每个个体。理论上联合应用16个STR位点,其个体的识别率可达0.999999999998。
所以当拿到自己新得的细胞系,或者传代很多的细胞系,不确定其是否存在污染时,就可以检测STR,通过与标准细胞库的细胞系STR图谱比较,从而确定细胞系的纯度。
鉴定STR的方法除了PCR外,还可以通过NGS的方法进行检测。
通过NGS数据进行检测的软件:
TSSV:https://pypi.org/project/tssv/
lobSTR:https://sourceforge.net/projects/lobstr/
STRait Razor:https://github.com/Ahhgust/STRaitRazor#documentation
HipSTR:https://github.com/tfwillems/HipSTR
STRSCan:http://darwin.informatics.indiana.edu/str/
以 HipSTR 为例:
git clone https://github.com/HipSTR-Tool/HipSTR
cd HipSTR
make
./HipSTR --help
安装完成后目录结构便是这样的。
如果出现软件的说明文档就说明安装成功了。
软件说明中数据是使用 “ Illumina whole-genome sequencing data ”,即 Illumina平台测序的全基因组数据,最好是在30X以上。
软件需要的输入文件是bam文件,可以使用--bams 参数输入各个bam文件,逗号隔开;或者使用--bam-files ,参数是包含所有bam的文件,一行一个bam文件。
--fasta 则是mapping时使用的参考基因组。
--regions 是关注的STR所在的bed区域。
--str-vcf 是VCF格式的输出文件结果。
hg19 基因组上STR bed文件:https://github.com/HipSTR-Tool/HipSTR-references/raw/master/human/hg19.hipstr_reference.bed.gz
hg38 基因组上STR bed文件:https://github.com/HipSTR-Tool/HipSTR-references/raw/master/human/hg38.hipstr_reference.bed.gz
bed文件格式如下图,第1-5列是必须的,分别是STR所在染色体,起始位置,终止位置,最小重复单元碱基数,重复次数。后边列为可选,分别是STR名称,以及序列。
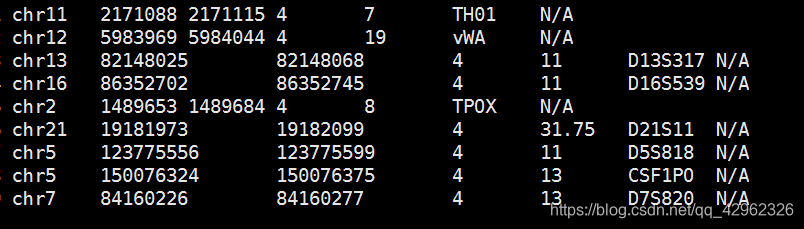
使用参数如下:
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
根据样本量的大小以及数据量的大小,可选参数:
--min-reads #进行分型时所需的最小reads数,当测序深度较低,reads较少时,可以通过改参数降低阈值(默认为100)
--max-str-len # STR序列的最大长度,大于该值时不被检测,如果一些STR较长,可以调节该参数
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
--min-reads 5
--max-str-len 300
软件在检测过程中,会自动使用 stutter-model (伪影模式?)的 EM algorithm 算法模型,对区域内的序列进行一些调整。如果样本,或者reads太少的话,用这个模型可能检测不出来,因为如果不符合EM algorithm所需的条件,这个位点就会被略过,这时可以使用--def-stutter-model 使用默认的模型。
./HipSTR --bams run1.bam,run2.bam,run3.bam,run4.bam
--fasta genome.fa
--regions str_regions.bed
--str-vcf str_calls.vcf.gz
--min-reads 5
--max-str-len 300
--def-stutter-model
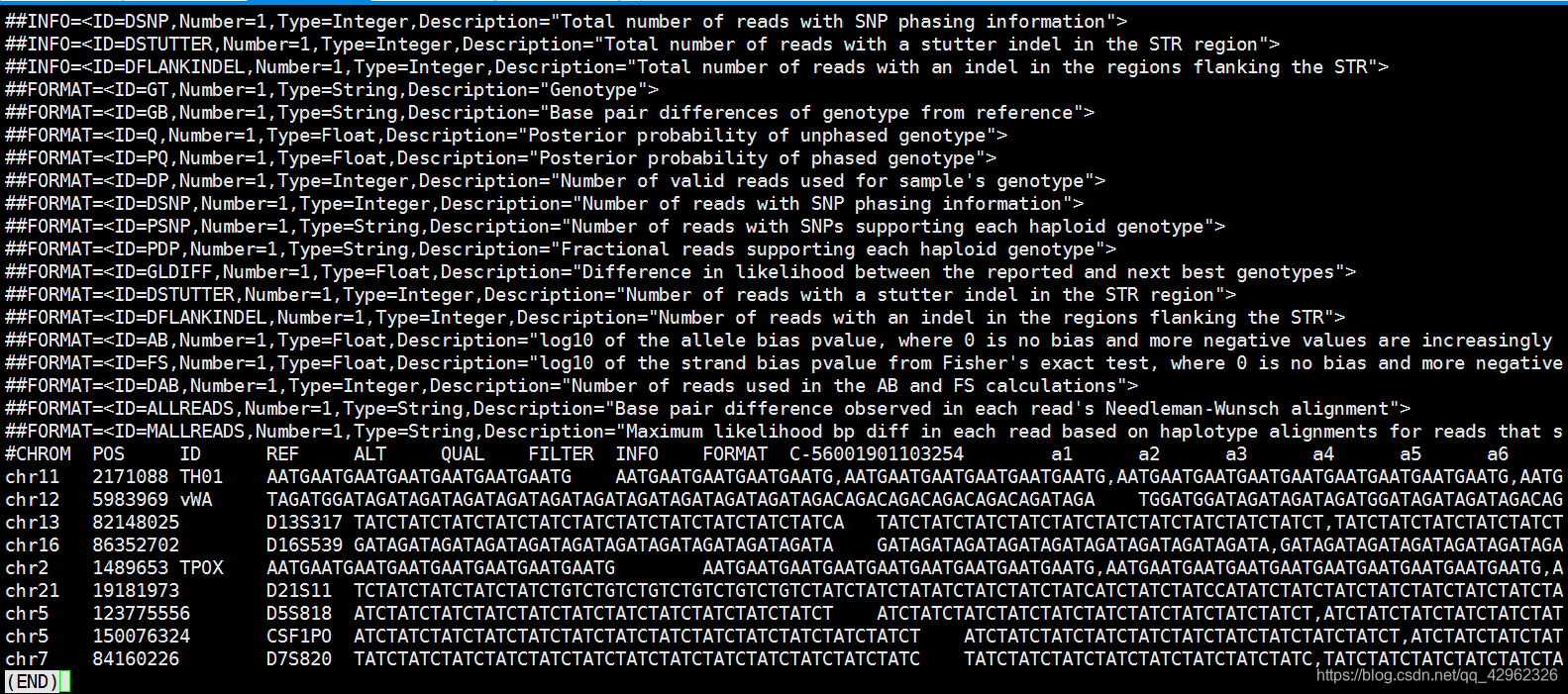
运行还是非常快的。输出结果为vcf格式文件。然后根据Ref, Alt ,以及info列的GT 就可以知道样本的STR基因型了, BPDIFFS 则是每个alt与ref的碱基差异数。
STR数据库: https://strbase.nist.gov
STR marker的序列信息特征:https://strbase.nist.gov/fbicore.htm
ATCC数据库:https://www.atcc.org/Products/Cells_and_Microorganisms/Testing_and_Characterization/STR_Profiling_Analysis.aspx
ATCC数据库细胞系STR查询向导文件:https://www.atcc.org/~/media/Documents/ATCC_STR_Database_Tutorial.ashx


















 568
568

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











