







Stringtie2——(2)参数
1.Attention!
1.Stringtie的输入文件必须按基因组位置排序。在STAR比对生成bam的过程中使用sorted…已经排好序

2.输入BAM文件中的每个 spliced read 比对(即跨越至少一个连接点的比对)必须包含标签XS,用以指示测序产生的read是来源于基因组序列上的哪条链产生的RNA。由TopHat和 HISAT2 (需参数 --dta,该参数用于发现剪接位点) 产生的比对结果中已经包含标签XS。——在STAR比对的过程中加参数!XS
一、基本用法
stringtie <aligned_reads.bam> [options]*
1.[options]
详见——>manual
- -j:连接点的覆盖度,即设置至少有这么多的spliced reads 比对到连接点(align across a junction)。 这个数字可以是分数, 因为有些reads可以比对到多个地方。 当一个read 比对到 n 个地方是,则此处连接点的覆盖度为1/n 。默认值为1。
- -s
- -f:将预测转录本的最低isoform的丰度设定为在给定基因座处组装的丰度最高的转录本的一部分。较低丰度的转录物通常是经加工的转录本的不完全剪接前体的artifacts。默认值为0.1。(0-1)
- -c:设置预测转录本所允许的最小read 覆盖度。 当一个转录本的覆盖度低于阈值,则输出文件中不含该转录本。默认值为 2.5
- -p:指定组装转录本的线程数(CPU)。默认值是1
- -A:输出基因丰度的文件(gene_abund.tab)
- -o:设置StringTie组装转录本的输出GTF文件的路径和文件名。此处可指定完整路径,在这种情况下,将根据需要创建目录。
- –merge:合并转录本。Stringtie将所有样本的GTF/GFF文件列表作为输入,将所有转录本合并/组装成飞冗余的转录本集合。可以用于生成一个跨多个RNA-seq样品的全局的、统一的转录本
二、使用
tailfix="Aligned.sortedByCoord.out"
for file in 'SRR11296675' 'SRR11296676' 'SRR11296677' 'SRR11296678' 'SRR11296679' 'SRR11296680' 'SRR11296681' 'SRR11296682'
do
echo $file$tailfix
stringtie /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/bbam/$file$tailfix.bam -j 2 -s 5 -f 0.05 -c 2 -p 5 \
-A /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/assembl/$file.gene_abund.tab \
-o /share2/pub/yangjy/yangjy/rna-seq-data/GSE146887/assembl/$file.out.gtf
done
parameter表示:需要至少 2 个读数用于连接组装,5 个读数用于单外显子转录物组装,每 bp 2 个读数用于多外显子转录物组装,5% 读数用于替代转录物组装。
三、输出文件
- GTF文件:记录组装的转录本信息
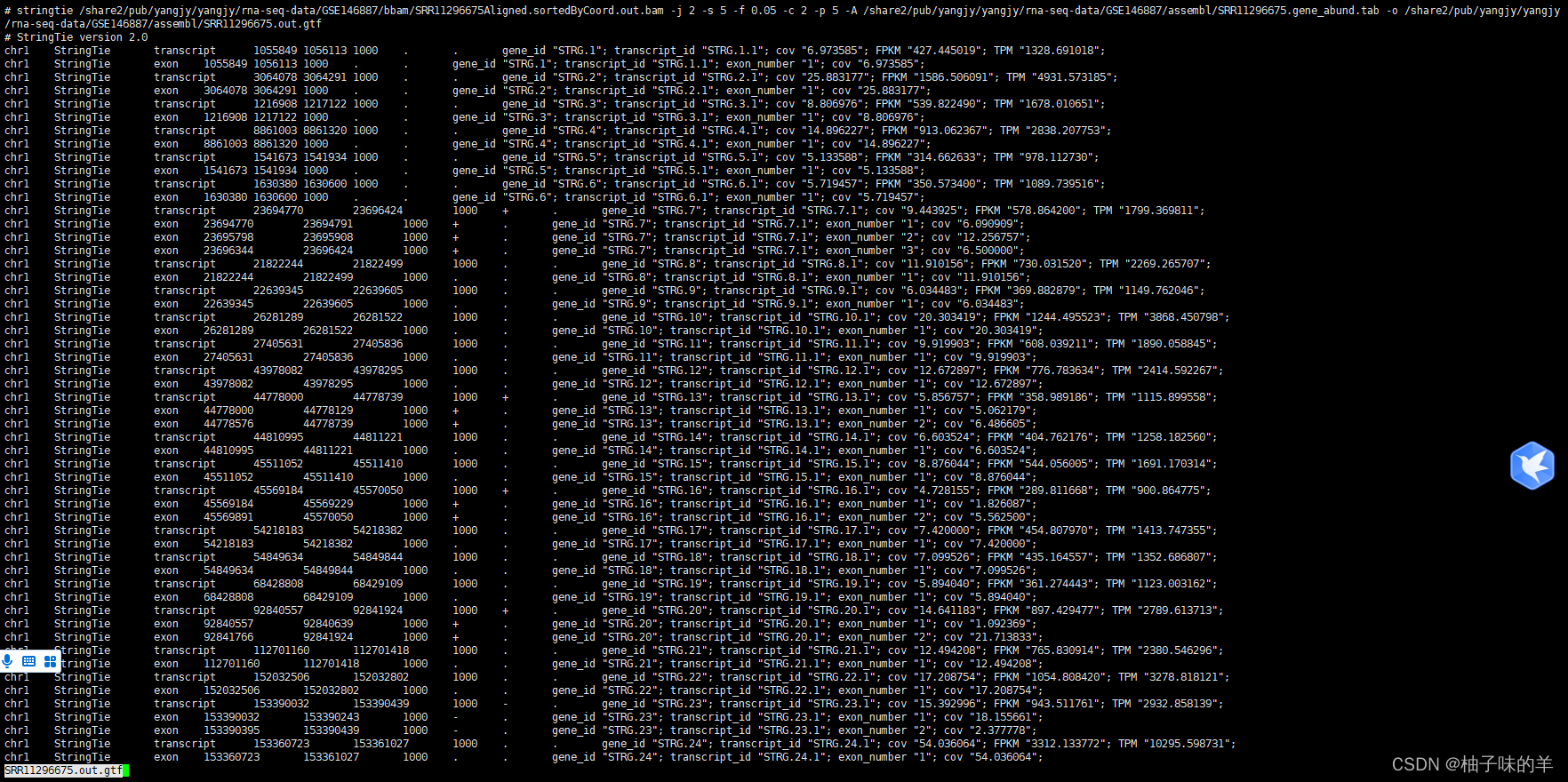
- Tab文件:记录基因丰度信息
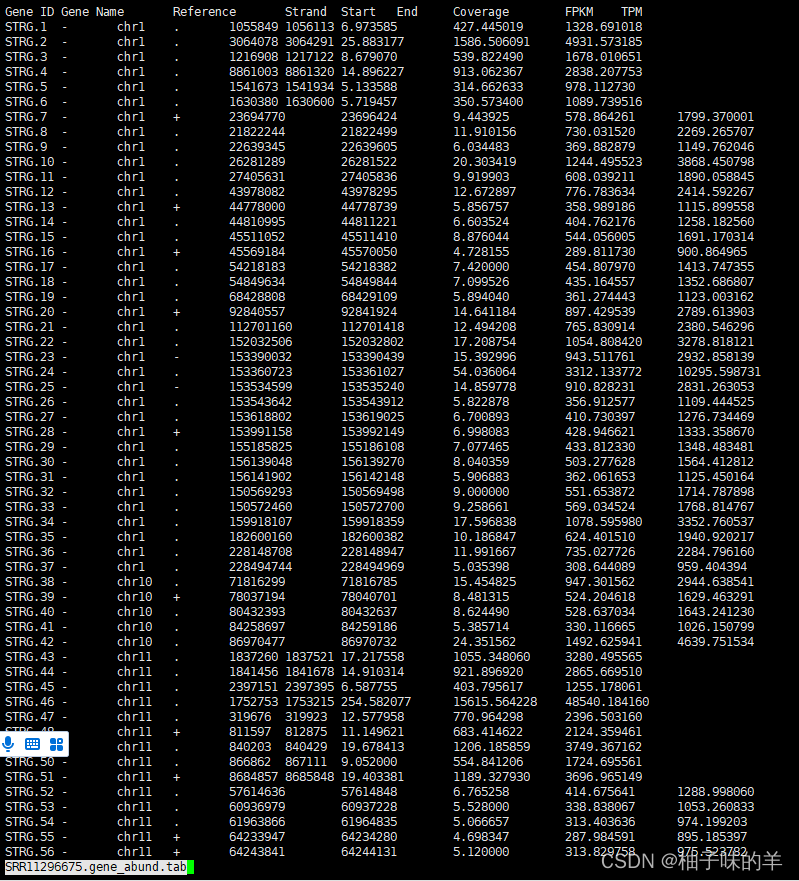
















 1436
1436











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











